
A class of linker free amphiphilic PEG grafted polymer support for linear and cyclic peptides†
M. A. Siyad and
G. S. Vinod Kumar*
Chemical Biology, Rajiv Gandhi Centre for Biotechnology, Thycaud P O. Thiruvananthapuram, 695014, India. E-mail: gsvinod@rgcb.res.in; Tel: +91 4712529526
First published on 29th October 2014
Abstract
PEG grafted (PEG400 and PEG1500), highly hydrophobic/hydrophilic balanced polymeric Schiff base dendron resins were synthesized by the reaction between diethylenetriamine and benzaldehyde molecules and characterized. The usefulness of the resins was demonstrated by synthesizing biologically significant linear as well as disulfide classes of endothelin peptides in high yields and purities.
Solid phase organic chemistry has evolved rapidly during the past few years mainly due to the enormous potential of peptide and non-peptide libraries in medicinal chemistry and chemical biology.1,2 The key factor for the success of solid phase synthesis is the right choice of polymeric support.3–6 Styrene and 4-chloromethyl styrene (VBC) copolymer cross-linked with divinylbenzene (DVB), commonly known as PS-DVB-VBC support, is the most commonly used support for solid phase peptide synthesis because of its good mechanical and chemical stability as well as the ease of derivatization of chlorine into a wide range of functional groups for substrate attachment. PS-DVB resin is completely hydrophobic in nature, whereas the growing peptide chain is much more hydrophilic. This difference induces a chain folding effect in which the peptide satisfies its own hydrogen bond requirements rather than being solvated. This can severely limit the synthetic access to the exposed end of the growing chains. These problems can be extensively rectified by increasing the hydrophilicity of the PS-DVB support.7
An easy and versatile approach that has emerged to improve the hydrophobicity of PS supports involves the replacement of DVB as the cross-linker in PS resins with relatively more flexible molecules.8,9 But it was a realized fact that cross-linking density has an optimum value beyond which produces negative effect in swelling character, an important parameter for solid support, due to restricted motion. It will severely limit the solvent imbibitions capacity too. An alternative approach is to graft short chain Polyethylene glycol (PEG) molecules on polystyrene backbone. Lengthening of ‘active group’ carrying chains to the reactive sites in which free mobility and reorganization of chains will result in high rate and fast completion of reaction known as spacer effect. But the grafting of PEG molecules reduces the functional loading abruptly. So we have adopted a better method by creating dendrimeric units up to the desired generation (G3) and incorporate hydrophilic moieties such as PEG to impart polar/hydrophilic nature. By this method, we have different choice of altering hydrophobic/hydrophilic balance such as (a) varying initial chlorine functional loading values (b) varying dendrimer generations (G1, G2, G3 etc.) (c) varying chain length of PEG units.
In order to achieve this, chemically robust dendritic sites have been generated up to third generation and subjected to PEGylation. Here we focused the on resin assembly of N,N-bis(ethylamine) dendrons by divergent route through Schiff base attachment up to the G3 generation. The Schiff base dendron was synthesized by the reaction between diethylenetriamine and benzaldehyde molecules. It was followed by PEG grafting (PEG400 and PEG1500) resulting in the formation of highly hydrophobic/hydrophilic balanced polymeric matrix. The utility of resins were demonstrated by synthesizing biologically significant linear as well as disulfide classes of endothelin peptides in high yields and purities. The beads of 200–400 mesh size were separated and 2 mol% VBC resin having chlorine value 0.068 mmol g−1 has been prepared and chosen for the entire studies such as dendrimer creation and PEGylation. Diethylenetriamine (dien) and aromatic aldehydes like benzaldehyde have been chosen for the preparation of dendron molecules because it readily forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Schiff base at room temperature and can be used directly without prior purification. The scheme (Scheme S1†) and 1H NMR data of Schiff base molecule obtained by the reaction between dien and benzaldehyde is given in Experimental section (ESI†).
:2 Schiff base at room temperature and can be used directly without prior purification. The scheme (Scheme S1†) and 1H NMR data of Schiff base molecule obtained by the reaction between dien and benzaldehyde is given in Experimental section (ESI†).
To the pre-swelled PS-DVB-VBC resin in 1,4-dioxane, excess quantity of Schiff base suspension has been added and subjected to heating at 100 °C for 48 h as shown in Scheme 1 as A1. The resin B1 gave characteristic IR absorption band at around 1645 cm−1 due to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the introduced Schiff base ligands.
N stretching vibration of the introduced Schiff base ligands.
 | ||
| Scheme 1 Synthesis of schiff base dendrimers to G3 generations. | ||
So the imine cleavage was done by acid hydrolysis using 6 M HCl followed by neutralization with 0.5 M NaOH solution resulting in resin C1 as shown in Scheme 1, having free amino groups. After acid hydrolysis, IR absorption peak at 1645 cm−1 disappears and new peak around 3400 cm−1 emerges, indicating the regeneration of primary amino groups. The amino loading capacities were quantitatively estimated by coupling with amino acid F-moc-Gly-OH followed by deprotection and absorbance measurement at 290 nm and obtained as 0.134 mmol g−1, i.e. around 97.06% of chlorine of chloromethyl styrene present in the backbone has been replaced by Schiff base units. We have preferred low initial capacity resin for dendrimer synthesis.
The diazotization reaction of aminated resin C1 at ice cold condition followed by neutralization with alkali forms hydroxyl resin D1 which is evidenced by shifting and broadening of the IR spectrum around 3500 cm−1. The hydroxyl loading capacity of D1 has been quantitatively analyzed by esterification using MSNT and F-moc-Gly-OH mixture. The hydroxyl capacity again verified by acylation reaction of hydroxyl groups with acetic anhydride–piperidine mixture and the acetic acid formed was back titrated with standard NaOH solution. Both quantifications gave concordant value of 0.133 mmol g−1 and the result obtained was in support with the amino loading value calculated. Thionyl chloride treatment of hydroxyl resin resulted in the formation of first generation (G1) chlorine terminated N,N-bisethylaminedendrimer resin E1, as shown in Scheme 1. The formation of chlorine was evidenced by the disappearance of hydroxyl peak in IR spectrum and loading was quantified by Volhard's estimation method as 0.134 mmol g−1 (Table 1).10 The chemical method for the preparation of B1, C1, D1 and E1 resins are shown in Scheme 2.
| Gen. | –NH2 (UV) mmol g−1 | –OH (UV) mmol g−1 | –OH (volumetric) mmol g−1 | –Cl (volumetric) mmol g−1 | % Conversion |
|---|---|---|---|---|---|
| G1 | 0.134 | 0.133 | 0.133 | 0.134 | 97.06 |
| G2 | 0.262 | 0.264 | 0.263 | 0.264 | 97.01 |
| G3 | 0.514 | 0.515 | 0.514 | 0.516 | 95.45 |
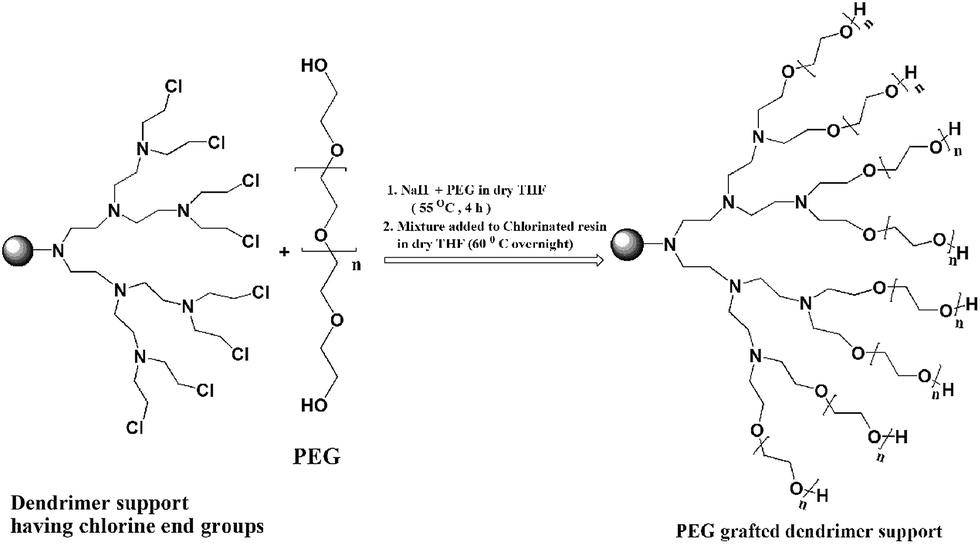 | ||
| Scheme 2 Method of PEG grafting to G3 dendrimer. | ||
In order to assess and confirm the percentage incorporation of Schiff base dendrons to G1, G2 and G3 dendrimers on PS-DVB-VBC, CHN analysis (data shown in ESI Table S1†) has been performed using amino ethyl G1, G2 and G3 resins (shown as C1, C2 and C3 in Scheme 1). In order to achieve the desired properties, we have selected poly(ethylene glycol) (PEG) as grafting material because PEG is chemically inert and robust which can impart hydrophilic character in to the polymer supports and are extensively exploited in different synthetic transformations.11 The optimum reaction conditions were achieved by grafting two different chain lengths of PEG having molar masses 400 Da and 1500 Da. The lyophilized chloro ethyl G3 dendrimer resins were independently subjected to Williamson's etherification with PEG molecules in dry THF under low pressure of N2 atm in an overnight reaction. The method of PEG grafting is shown in Scheme 2.
The FTIR spectrum of both PEG400 and PEG1500 grafted systems showed broad intense peaks around 1100 cm−1 and 3500 cm−1 corresponding to the ethereal C–O–C linkage and free hydroxyl groups of PEG. The IR spectra of different chemical transformations are summarized in ESI (Fig. S1†). The hydroxyl loading capacities of resins after PEG grafting were quantified by UV-Visible and volumetric methods. It has been observed that after PEG grafting, the hydroxyl loading values were found to decrease drastically and the values obtained were 0.203 and 0.326 mmol g−1 for PEG1500 and PEG400 respectively. The integrity of the polymer and PEG grafting has been further evidenced by solid state 13C NMR (ESI Fig. S2a†) and surface morphological changes using scanning electron microscopy. PEG grafting is also supported by the SEM images in which the smooth and clean surface of initial PS-DVB-VBC support become rough, irregular and bumpy because of the extensive incorporation of PEG molecules into the polymer matrix (shown in ESI Fig. S2b†). The PEG1500 grafted G3 support was selected for checking the chemical stability studies and was tested in various Fmoc and Boc peptide synthetic conditions.
The G3 dendrimer support having hydroxyl end groups was used to check the swelling nature and compared to PEGylated dendrimer supports. It was observed that hydroxy terminated G3 dendrimer matrix showed better swelling characteristics than commercially available PS-DVB support and PEG1500 grafted dendrimeric support displayed better swelling nature compared to PEG400, especially in polar solvents, and it might be due to long PEG chains which will impart high hydrophilic character to the resin and will enhance easy uptake of solvents into the polymer matrix. Swelling characteristics are depicted in ESI Fig. S3.†
Peptide 1: linear
In order to ensure the efficiency of both resins towards various coupling and deprotection reactions used in peptide synthesis, a highly selective ligand for ETB receptor, i.e. ETB receptor agonist having 21 amino acid residue has been synthesized on both PEG400 and PEG1500 grafted dendrimer resins and compared the yield and purity. The peptide having amino acid sequence H2N-Ala-Ser-Ala-Ser-Ser-Leu-Met-Asp-Lys-Glu-Ala-Val-Tyr-Phe-Ala-His-Leu-Asp-Ile-Ile-Trp-OH (peptide 1) was selected as model peptide for comparison. Since PEG having free hydroxyl end groups, the carboxyl group of C-terminal tryptophan can attach directly to the resin via an ester bond using MSNT in dry DCM without a linker.The standard Fmoc strategy has been followed throughout the synthesis to build the peptides on both supports in identical synthetic conditions. In most cases of amino acid acylation and deprotection reactions, the number of couplings as well as the time taken for completion of reaction has been found to be reduced for PEG grafted resins compared to commercial available Merrifield resin. Among PEG grafted supports, PEG1500 grafted resin showed better efficiency towards minimum reagent availability and enhanced rate of reactions in all stages of synthesis. The peptides after synthesis were detached from the supports using the cleavage cocktail comprising of 94% TFA: 2.5% water: 2.5% EDT: 1% TIS. The yields of the crude peptides obtained after cleavage were 91% and 84% for PEG1500 and PEG400 respectively. HPLC results of peptides synthesized using PEG400 and PEG1500 are summarized in Fig. 1a and c. The linear peptide synthesized on PEG1500 grafted support showed comparatively better yield and purity with respect to PEG400. The syntheses were confirmed by MALDI-TOF analysis and results are shown in Fig. 1b and d for PEG400 and PEG1500 respectively.
 | ||
| Fig. 1 ETB receptor agonist synthesized on PEG400 (a) HPLC (b) corresponding MALDI-TOF; ETB receptor agonist synthesized on PEG1500 (c) HPLC (d) MALDI-TOF. | ||
Peptide 2: single cyclic bond
Using the newly developed PEG grafted supports, we have synthesized biologically potent ETB receptor antagonist having single disulfide bonds between 1st and 5th cysteine residues (peptide 2). The air oxidation has been conducted on PEG400 support and TFA–DMSO–anisole method has been performed on PEG1500 resin. The amino acid sequence of ETB receptor antagonist having single disulphide bonds between 1st and 5th positions are shown in Scheme 3. | ||
| Scheme 3 ETB receptor antagonist synthesised on PEG400 & PEG1500. | ||
Air oxidation method has been performed on PEG400 grafted resin and oxidation of peptides to form cyclic disulphide bridges are achieved by treating dilute solution of peptides at weakly basic pH values. The peptide cleaved from the support is in reduced sulfhydryl form first which will be readily converted to cyclic disulphide bonded forms by air oxidation. Here the disulphide bridge was formed between 1st and 5th positions and hence cys amino acids having trityl (Trt) as thiol protecting groups has been selected and recommended, as it is labile to TFA and can be easily removed during normal TFA cleavage procedure. The completion of disulfide bond formation is monitored by Ellman's method.12 The HPLC diagrams of peptides before and after air oxidation is shown in Fig. 2a and b and corresponding masses by MALDI-TOF is shown in Fig. 2c and d. From the HPLC chromatogram, it is clear that after disulphide bond formation, elution time has been shifted to lower value. The disulfide bridge formation was again proved from MALDI TOF analysis. The peptide after cleavage (before air oxidation) showed a peak at M/Z = 1411.254 corresponding to the reduced sulfhydryl form and after air oxidation the peak at M/Z = 1409.233 corresponding to the oxidized disulfide bonded form. The HPLC chromatogram after air oxidation showed small peaks in addition to main peak which might be of the formation of fully folded or partially folded peptides. The undesired products were formed mainly because of polymeric inter chain disulphide bond formation occurred in addition to desired intra chain formation while the course of reaction. The yield of the crude peptides before and after air oxidation was 91% and 88% respectively. The crude peptide obtained after air oxidation was further purified by preparative HPLC and the pure peptide collected was 66% yield.
 | ||
| Fig. 2 HPLC profiles of ETB receptor antagonist synthesized on PEG400(a) before air oxidation (b) after air oxidation; MALDI-TOF analysis (c) before air oxidation (d) after air oxidation. | ||
The disadvantage of air oxidation process is that reactions can sometimes be very slow and low yielding. The polymeric undesired inter chain disulfide bond formation by air oxidation method can be minimized to certain extent by utilizing the TFA–DMSO–anisole oxidation method13 to form region selective disulfide peptides. Using PEG1500 grafted support, we have synthesized the same ETB receptor antagonist peptide (peptide 2) by TFA–DMSO–anisole method. For that we have used cysteine residues in which S-atom was protected with t-butyl (t-Bu) group which will rapidly cleave and convert to cystine at room temperature in presence of TFA–DMSO–anisole. After cleavage, 10 mg of crude peptide was dissolved in 10 mL of TFA–DMSO–anisole (97.9:2:0.1, v/v) in a 50 mL round bottom flask and the mixture was stirred for 1 h at room temperature. The excess TFA was then removed in vacuum and the peptide was precipitated by the addition of chilled ether. The purity of the peptides before and after oxidation was checked with HPLC and masses by MALDI-TOF. The yields of the crude peptides obtained after cleavage and after TFA–DMSO–anisole oxidation were 92% and 79% respectively. The crude product after oxidation was further purified by preparative HPLC yielding the peptide in 62% yield. Analytical HPLC data for the t-Bu protected peptide after cleavage from the support, the crude peptide after TFA–DMSO–anisole oxidation and purified product are shown in Fig. 3a–c respectively. MALDI-TOF analysis of the peptide solution revealed that the cleavage of the t-Bu groups and oxidation of the disulfide is completed. The MALDI-TOF results before and after TFA–DMSO–anisole oxidations are shown in Fig. 3d and e.
 | ||
| Fig. 3 HPLC profiles of ETB receptor antagonist synthesized on PEG1500 (a) before TFA–DMSO–anisole oxidation (b) after TFA–DMSO–anisole method (crude) (c) purified; MALDI-TOF analysis (d) before TFA–DMSO–anisole oxidation (e) after oxidation. | ||
Peptide 2: double cyclic bond
Using PEG1500 grafted support, biologically significant endothelin-1 human peptide having double disulfide structure was synthesized and analyzed. Human endothelin-1 (hET-1) is a 21 amino acid peptide having two intra molecular disulfide bonds between Cys1–15 and Cys3–11 having a C-terminal tryptophan residue. Its double disulfide structure and C-terminal Trp are essential for the vasoconstrictor activity of hET-1. Endothelin-1, human peptide having 21 amino acid sequence and double disulfide structure is shown in Scheme 4 (peptide 3). | ||
| Scheme 4 hET-1 synthesized on PEG1500. | ||
To construct the disulfide bonds region selectively, combination of successive oxidation with air and TFA–DMSO–anisole using trityl (Trt) and t-Bu (t-butyl) as orthogonal thiol protecting groups were employed. The 3rd and 11th positions were occupied by Cys(Trt) group which is labile in TFA and 1st and 15th positions were occupied by Cys(t-Bu) group which is stable in TFA and kinetically labile in TFA–DMSO–anisole. We have constructed the peptide chain by the standard Fmoc method starting from C-terminal tryptophan using MSNT and 1-MeI in dry DCM. The trityl groups present in 3rd and 11th positions were labile to TFA and consequently removed during the normal course of the cleavage reaction along with other side chain protecting groups present in peptide chain except the tert-butyl groups present in 1st and 15th positions which are stable enough to resist the TFA attack. Due to the high stability of tritylcation and the strong nucleophilic nature of thiol groups, removal of trityl reaction was reversible and so special care needs to be given to the cleavage conditions to ensure complete deprotection.
This problem can be overcome by employing cleavage cocktail which contains triisopropylsilane. This reagent is extremely effective at quenching the tritylcation, converting it irreversibly to triphenyl methane. Finally the peptide was removed from the support using the cleavage cocktail having TFA:water:EDT:TIS in volume ratio of 94:2.5:2.5:1. After cleavage the sulfur atom of cysteine amino acids present in 3rd and 11th positions will exist as reduced sulfhydryl forms which can be readily oxidized by atmospheric oxygen by exposing the peptides in air in weakly basic medium. So after washing with cold ether, some portion of the peptides were removed and kept in a freeze dried condition and stored under dry N2 atm. for HPLC analysis. The remaining portion of peptide was subjected to air oxidation by dissolving the peptides in deaerated ammonium bicarbonate solution and left the mixture to stand open in atmosphere until the reaction was completed.
In order to avoid the polymeric chain formation, low concentration of linear peptide solution was used for air oxidation. The completion of the reaction was monitored by Ellman test. The single disulfide peptide was lyophilized and analyzed by HPLC. The HPLC of peptides after cleavage and after air oxidation are shown in Fig. 4a and b. The shifting of main peak in HPLC chromatogram after air oxidation to the lower elution time indicates the disulfide bond formation. The synthesis was again confirmed by observing the masses of peptides before and after air oxidation by MALDI-TOF analysis and results are summarized in Fig. 4e and f. The yield of the crude peptide obtained after cleavage from the support and after air oxidation was 93% and 86% respectively. The air oxidized peptide was further treated for second disulfide bond formation by reacting with TFA–DMSO–anisole in volume ratio 97.9:2:0.1, v/v). Air oxidized peptide (10 mg) was dissolved in 10 mL of reaction cocktail containing TFA–DMSO–anisole (97.9:2:0.1, v/v) in 100 mL round bottom flask and the mixture was stirred occasionally with magnetic beads for one hour at room temperature. The excess volume of TFA was removed under reduced pressure and the peptide was further precipitated by the addition of cold ether. MALDI-TOF analysis of the peptide solution revealed that the cleavage of the t-Bu groups and oxidation of the disulfide was completed. The yield of the crude peptide obtained after TFA–DMSO–anisole oxidation is 76%. The crude peptide further purified by preparative HPLC yielding the desired peptide is 62% yield. Fig. 4c and d showed the analytical HPLC data for the crude fully folded disulfide bonded peptide and the purified final product. The accurate mass analysis after oxidation was also performed using MALDI-TOF to confirm the synthesis as shown in Fig. 4g.
 | ||
| Fig. 4 HPLC profiles of hET-1 peptide synthesized on PEG1500 (a) before air oxidation (b) after air oxidation (c) after TFA–DMSO–anisole oxidation (crude) (d) corresponding purified; MALDI-TOF analysis (e) before air oxidation (f) after air oxidation (g) after TFA–DMSO–anisole oxidation. | ||
Conclusions
Novel classes of low and high molecular weight amphiphilic linker free PEG grafted polymer supports were developed to facilitate easy penetration of molecules/solvents into the macromolecular matrices. PS-DVB-VBC resin was used for dendrimer creation because of its high mechanical, chemical and thermal stoutness and the facility of derivatization to different functional groups for a wide range of substrate attachments. Low initial functional loading support was preferred to avoid all the difficulties customarily encountered due to steric hindrances associated with the bulky nature of Schiff base molecules and crowded and closely packed polystyrene back bone. Biologically potent endothelin classes of linear as well as single and double disulfide bonded peptides are synthesized effectively and conveniently on both PEG grafted supports by air and TFA–DMSO–anisole methods in high yield and purity and proved as an ideal support for solid phase synthesis of peptides and peptide mimetics. These results presented here demonstrate the use of newly developed PEG grafted poly(N,N-bisethylamine) dendrimer resins that will translate well for applications to solid phase synthesis and combinatorial libraries currently being investigated in our laboratory.Notes and references
- K. S. Lam, S. E. Salmn, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, Nature, 1991, 354, 82–84 CrossRef CAS PubMed.
- H. R. Rengifo, C. Grigoras, B. I. Dach, X. Li, N. J. Turro, H. Lee, W. Wu and J. T. Koberstein, Macromolecules, 2012, 45, 3866–3873 CrossRef CAS.
- Y. Wang and M. D. Distefano, Chem. Commun., 2012, 48, 8228–8230 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2153 CrossRef CAS.
- M. Hurevich and P. H. Seeberger, Chem. Commun., 2014, 50, 1851–1853 RSC.
- M. A. Siyad, A. S. V. Nair and G. S. V. Kumar, J. Comb. Chem., 2010, 12, 298–305 CrossRef CAS PubMed.
- M. A. Siyad and G. S. V. Kumar, Biopolymers, 2012, 98, 239–248 CrossRef CAS PubMed.
- M. A. Siyad and G. S. V. Kumar, Comb. Chem. High Throughput Screening, 2012, 15, 386–394 CrossRef CAS.
- M. A. Siyad and G. S. V. Kumar, Curr. Org. Synth., 2013, 15, 318–327 CrossRef.
- J. M. Stewartand and J. D. Young, Solid Phase Peptide Synthesis, PierceChemical Company, Rockford, 1L, 1984 Search PubMed.
- Z. Wang and R. L. Yang, Sci. China: Chem., 2010, 53, 1844–1852 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS.
- A. Cuthberston and B. Indrevoll, Tetrahedron Lett., 2000, 41, 3661–3663 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08020c |
| This journal is © The Royal Society of Chemistry 2014 |