
The convenient aqueous synthesis and biological evaluation of ortho-(3,4,5-trimethoxybenzoyl)-acetanilides as novel anti-cancer agents†
Abstract
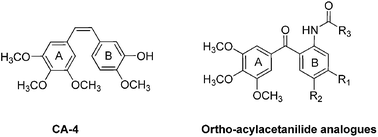
A series of new ortho-(3,4,5-trimethoxybenzoyl)-acetanilides were synthesised by the cross-coupling reaction catalyzed with Pd catalyst in aqueous medium, with polyethylene glycol as additive under very mild conditions. The evaluation of these compounds as tubulin polymerization inhibitors indicated that most of these compounds were potential anti-cancer agents. Among them, compound 13, 2-hydroxy-N-(5-methoxy-2-(3,4,5-trimethoxy benzoyl)phenyl) acetamide exhibited excellent antiproliferative activity against various human cancer cell lines (GI50 = 71 nM for human HeLa cell line), and good activity for inhibiting tubulin polymerization (IC50 = 2.94 μM). The mechanism study indicated that compound 13 could arrest cell-cycle progression at the mitosis phase, block the formation of cdc2/cyclin B1 complex, down-regulate the p-Cdc 25C and the anti-apoptotic proteins Bcl-2 and Bcl-XL, and up-regulate the pro-apoptotic proteins Bax and Bad.
 Please wait while we load your content...
Please wait while we load your content...

