
Continuous-flow azide–alkyne cycloadditions with an effective bimetallic catalyst and a simple scavenger system†
Sándor B. Ötvösa,
Gábor Hatossa,
Ádám Georgiádesa,
Szabolcs Kovácsb,
István M. Mánditya,
Zoltán Novákb and
Ferenc Fülöp*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, 6720 Szeged, Hungary. E-mail: fulop@pharm.u-szeged.hu
bMTA-ELTE “Lendület” Catalysis and Organic Synthesis Research Group, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter stny. 1/a, Budapest, 1117, Hungary
First published on 15th September 2014
Abstract
A flow chemistry-based technique is presented herein for Cu(I)-catalyzed azide–alkyne cycloadditions with a copper on iron bimetallic system as the catalyst and iron powder as a readily available copper scavenger. The method proved to be rapid and safe as compared with the conventional batch experiment; and by using an in-line copper scavenger, the level of copper impurities in the triazole products could readily be reduced to negligibly small amounts. The process was widely applicable, as not only terminal alkynes, but also various disubstituted acetylenes were nicely tolerated as dipolarophiles leading to useful 1,4,5-trisubstituted 1,2,3-triazoles.
Introduction
The 1,3-dipolar cycloaddition of organic azides with alkynes as dipolarophiles is the most straightforward way to obtain useful 1,2,3-triazoles.1 The classical Huisgen reaction utilizes only thermal induction and gives an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1,4- and 1,5-disubstituted 1,2,3-triazoles,2 but with Cu(I) catalysis the 1,4-disubstituted isomer is formed regioselectively.3 Applications of the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) have contributed to many areas of modern chemistry, such as bioconjugation, peptidomimetic chemistry, polymer and materials sciences and supramolecular chemistry.4 As a consequence of the potential biological properties of 1,2,3-triazole-containing compounds, the CuAAC reaction has recently become a versatile tool for the drug discovery.5
:1 mixture of 1,4- and 1,5-disubstituted 1,2,3-triazoles,2 but with Cu(I) catalysis the 1,4-disubstituted isomer is formed regioselectively.3 Applications of the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) have contributed to many areas of modern chemistry, such as bioconjugation, peptidomimetic chemistry, polymer and materials sciences and supramolecular chemistry.4 As a consequence of the potential biological properties of 1,2,3-triazole-containing compounds, the CuAAC reaction has recently become a versatile tool for the drug discovery.5
As a novel sustainable alternative for conventional batch-based synthetic techniques, continuous-flow (CF) processing has recently emerged in academic and industrial research of fine chemicals.6 CF-based approaches offer significant advantages over classical segmented unit operations,7 such as enhanced heat and mass transfer and improved mixing properties,8 increased parameter space with an unprecedented level of control over the most important reaction conditions,9 and inherently safer and greener chemistry and higher reaction rates.10
The synthesis of 1,2,3-triazoles via CuAAC comprises an extraordinarily robust and straightforward transformation, which can be performed under a wide array of reaction conditions.11 For such reactions, the safety aspects associated with the handling of explosive azides,12 the inherent scalability of continuous processing and the plausible opportunity for automation have made CF CuAAC approaches particularly appealing.13 In CF reaction technology, mainly heterogeneous approaches, such as copper-in-charcoal (Cu/C),14 immobilized Cu(I) complexes,15 copper powder16 or heated copper tubing17 are employed as sources of catalytically active copper species. Despite using a heterogeneous catalyst, metal impurities often reduce the organic purity of the triazole product due to significant leaching of copper from the catalytic source.14b Flow chemistry enables various in-line techniques (such as the use of metal scavenger resins or CF extraction processes) to reduce copper contamination and to obtain sufficiently pure products with less purification and work-up steps.15b,18
Herein, we report an effective CF technique for CuAAC reactions employing the combination of a copper on iron (Cu/Fe) bimetallic catalytic system and iron powder as a readily available copper scavenging unit, which can practically act as an in situ generated catalyst after several hours of continuous operation as a scavenger. The synthetic capabilities of the CF process were validated with the synthesis of a series of 1,4-disubstituted and 1,4,5-trisubstituted 1,2,3-triazoles.
Experimental
CF methodology
Experiments were conducted in a CF reactor system shown in Fig. 1. A stainless steel cartridge (with internal dimensions of 70 × 4 mm) was employed as the catalyst bed, which was placed in a heating unit or an ultrasonic bath (with a power of 150 W). The catalyst bed was combined with a coiled stainless steel line (with an internal diameter of 500 μm) for preheating of the liquid phase before entering the column. A backpressure regulator ensured constant pressures up to a maximum of 100 bar, and the mixture of the reactants was pumped through the system continuously by means of a conventional HPLC pump. For reactions with in line copper scavenging, a second cartridge (with the same dimensions) was filled with scavenger and fitted into the flow line. | ||
| Fig. 1 Experimental setup for the CF reactions. | ||
Materials
Copper powder (average particle size: 200 μm) and 5 wt% Cu/C (average particle size: 20 μm) were purchased from Aldrich, and used as received. 5 wt% Cu/Fe (average particle size: 200 μm) was prepared from CuSO4 and iron powder as reducing agent, according to the previously described procedure.19 Iron powder (used as scavenger) was purchased from Alfa Aesar with an average particle size of 200 μm. Azides, as starting materials were prepared with literature procedures in purities of >99%, and were therefore used without further purification.16a,19b,20General procedure
In the case of copper powder and Cu/Fe, 1 g of the heterogeneous catalyst was filled into a catalyst bed, respectively. Due to the large porosity of activated charcoal, it was possible to load only 0.5 g of Cu/C into the cartridge at a maximum. The scavenger column was loaded with 1 g of iron powder.For the small-scale CF reactions, 0.84 mmol (1 equiv.) of the azide and 1.26 mmol (1.5 equiv.) of the alkyne were dissolved in 10 mL CH2Cl2. If necessary, 0.0336 mmol (0.04 equiv.) of N,N-diisopropylethylamine (DIEA) and 0.0336 mmol (0.04 equiv.) of glacial acetic acid (AcOH) were added as additives. After homogenization, the solution was pumped through the CF reactor under the appropriate conditions. Between two CF reactions the catalyst bed and the scavenger were washed at room temperature (RT) for 10 min with CH2Cl2 at a flow rate of 1 mL min−1. The concentration of the reactants was not varied upon scaling-up, and a larger amount of starting medium was simply pumped through the instrument. The crude products were checked by thin-layer chromatography with a mixture of n-hexane–EtOAc as eluent, and the solvent was next evaporated off under vacuum. If necessary, column chromatographic purification was carried out on silica gel with a mixture of n-hexane–EtOAc as eluent.
To determine the residence time on the filled catalyst bed, a solution of a dye was pumped through the cartridge. The time that elapsed between the first contact of the dye with the catalyst bed and the moment when the colored solution appeared at the column output was measured.
Product analysis
The obtained 1,2,3-triazoles were characterized by NMR experiments and elemental analyses. 1H and 13C NMR spectra were recorded on a Bruker Avance DRX 400 spectrometer, in CDCl3 as solvent, with TMS as internal standard, at 400.1 and 100.6 MHz, respectively. Microanalyses were performed on a Perkin-Elmer 2400 elemental analyzer.The copper concentration of the samples was determined by inductively coupled plasma mass spectrometer (ICP-MS), using an Agilent 7700x-type instrument equipped with a collision cell after digestion with concentrated HNO3. The determination was carried out on the isotope 63Cu, with He as collision gas.
The morphology of the catalyst and the scavenger was examined with scanning electron microscopy (SEM – Hitachi S-4700 microscope with varying acceleration voltage). The samples were fixed on a double-sided adhesive carbon tape and were coated with gold using a sputter coater (Quorum Technologies SC7620). The approximate composition and the elemental map of the substances were investigated by a Röntec QX2 energy dispersive X-ray fluorescence spectrometer (EDX) coupled to the microscope.
Results and discussion
Cu/Fe is a versatile bimetallic catalyst system which consists of nano-sized copper particles deposited onto an iron surface (Fig. 2).19,21 It can easily be prepared from Cu(II) salts and iron powder which serves as a non-toxic reducing agent and a support for copper particles at the same time. As the beginning of our study, we explored the merits of the Cu/Fe system for CuAAC reactions in a high-pressure CF device in comparison with Cu/C and copper powder as commercially available Cu(I) sources and also with literature batch data employing the same catalyst (Table 1). The 1,3-dipolar cycloaddition between benzyl azide and phenylacetylene was chosen as a test reaction with CH2Cl2 as solvent. A reaction mixture containing 1 equiv. of the azide and 1.5 equiv. of the alkyne was pumped through the reactor in each run. The azide was applied in a concentration of 0.085 M, which was the highest possible concentration preventing the precipitation of the resulting product and a blockage in the channels of the reactor. All CF reactions were carried out at a flow rate of 0.5 mL min−1 which involved a residence time on the catalyst bed as low as 1.5 min, and thus a prominently short process time. | ||
| Fig. 2 SEM image of the Cu/Fe catalyst. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | p (bar) | T (°C) | Additives | Conversion of 1a (%) | |
| DIEA (equiv.) | AcOH (equiv.) | |||||
| a Determined by 1H NMR spectroscopic analysis of the crude material.b Batch reference data.19b Reaction conditions: azide (0.5 mmol, 1 equiv.), acetylene (0.5 mmol, 1 equiv.), Cu/Fe (5 mol%, 5 wt%) (32 mg, 0.025 mmol copper), CH2Cl2, (300 μL), stirring at 30 °C for 8 h.c US stands for ultra sound.d Isolated yield. | ||||||
| 1 | Copper powder | 1 | RT | 0 | 0 | 19 |
| 2 | Copper powder | 100 | RT | 0 | 0 | 31 |
| 3 | Copper powder | 100 | 100 | 0 | 0 | Quant. |
| 4 | Copper powder | 100 | RT | 0.04 | 0.04 | Quant. |
| 5 | Copper powder | 100 | RT/USc | 0 | 0 | 54 |
| 6 | Cu/C | 1 | RT | 0 | 0 | 6 |
| 7 | Cu/C | 100 | RT | 0 | 0 | 11 |
| 8 | Cu/C | 100 | 100 | 0 | 0 | 38 |
| 9 | Cu/C | 100 | RT | 0.04 | 0.04 | 81 |
| 10 | Cu/C | 100 | RT/USc | 0 | 0 | 15 |
| 11 | Cu/Fe | 1 | RT | 0 | 0 | 21 |
| 12 | Cu/Fe | 100 | RT | 0 | 0 | 28 |
| 13 | Cu/Fe | 100 | 100 | 0 | 0 | Quant. (98)d |
| 14 | Cu/Fe | 100 | RT | 0.04 | 0.04 | Quant. (99)d |
| 15 | Cu/Fe | 100 | RT/USc | 0 | 0 | 49 |
| 16b | Cu/Fe | 1 | 30 | 0 | 0 | Quant. (98)d |
At ambient temperature and pressure, low conversion was obtained with each catalyst (entries 1, 6 and 11), and thus various approaches were evaluated to improve the reactivity. High pressures can enhance the rate of the triazole formation in accordance with Le Chatelier's principle, and accordingly we found higher conversions at 100 bar (entries 2, 7 and 12).16a Elevation of the pressure allowed the use of temperatures well above the boiling point of CH2Cl2, thus, as a next step, high-temperature conditions were examined. We had previously concluded that the rate of the triazole formation is highly dependent on the temperature, and heating to at least 100 °C was necessary to obtain quantitative conversion with copper powder as catalytic source.16a At 100 bar and 100 °C Cu/Fe and copper powder performed equally well, as quantitative conversion was achieved in both test reactions (entries 3 and 13). In contrast, with Cu/C a conversion of only 38% was found (entry 8). However due to the risks associated with the handling of explosive azides it is highly beneficial to improve the rates of the CuAAC reactions without high temperatures. Amines, as basic additives, are known to boost the reactivity of the CuAAC considerably,3a,11,22 and it was recently shown that the addition of catalytic amounts of certain acids can further accelerate the triazole formation and also prevent the accumulation of unwanted byproducts.23 To realize the CF CuAAC at ambient temperature, we utilized 0.04 equiv. of DIEA and 0.04 equiv. of AcOH jointly as additives.16a We were pleased to find that the performance of the Cu/Fe catalyst was comparable with copper powder, and quantitative conversion was achieved with additives in both cases (entries 4 and 14). In case of Cu/C the use of additives nicely improved the reactivity as compared with the high-temperature/high-pressure conditions, and gave an acceptable conversion of 81% (entry 9). We also checked the effects of ultrasound promotion to relieve harsh reaction conditions.24 For this, the filled catalyst column was simply placed into an ultrasonic bath. It was found that sonication improved the rates of the CF reaction at ambient temperature in case of the bimetallic Cu/Fe system and copper powder, but the efficacy of this approach fell behind the use of additives, as a conversion of only ∼50% was obtained with both catalysts (entries 5 and 15). In case of Cu/C, not more than a negligible conversion increase was found upon sonication (entry 10). The overall lower reactivity of Cu/C may be explained with the poorer accessibility of the catalytically active Cu(I) species in the activated charcoal matrix and with the lower catalyst loading in the cartridge than is the case of Cu/Fe and copper powder.
The CF results with the Cu/Fe catalyst system (obtained either under high-temperature/high-pressure conditions or under ambient temperature with additives) compare well with the corresponding literature batch data utilizing the same catalyst and conventional mechanical stirring at 30 °C (entries 13, 14 vs. 16).19b It should be pointed out that the batch reaction required 8 h of reaction time for completion, whereas the continuous process offered quantitative conversion in a short residence time on the catalyst bed, and thus it proved to be much more rapid. Another point is that the CF method tends to be safer due to the contained environment, the lack of headspace and the well-defined short residence time.
In reactions involving heterogeneous catalysts, leaching of copper from the catalyst bed can reduce the purity of the triazole products and necessitate further work-up and purification steps. In our CF reactions, trace amounts of copper were determined by means of ICP-MS measurements in the quantitatively obtained triazole products (Table 2) without any work-up and purification steps other than a simple evaporation (except for entry 3). The analytical data show that the copper content of the crude products obtained under high-pressure/high-temperature conditions without any additives were lower than those when DIEA and AcOH were jointly applied (entries 1 vs. 2). This is because amines, as basic additives, can coordinate to the catalytically active Cu(I) species and promote their liberation from the copper matrix, thereby increasing the copper content of the crude product. In spite of this fact, the utilization of additives for rate enhancement in the CF CuAAC is much wiser than the use of harsh reaction conditions due to safety considerations. Thus, to reduce metal contamination of the triazole product an in-line copper scavenging methodology is highly beneficial.
|
||
|---|---|---|
| Entry | Description of the reaction conditionsa | Copper contentb,c (μg g−1) |
| a Unused portions of the catalyst and the scavenger were used for each reactions.b Determined by ICP-MS.c 10 mL aliquots of the reaction mixture was pumped through the system for each ICP-MS samples.d Additives: 0.04 equiv. DIEA + 0.04 equiv. AcOH. | ||
| 1 | Cu/Fe, 100 bar, 100 °C, without additives | 41.0 ± 0.8 |
| 2 | Cu/Fe, 100 bar, RT, with additivesd | 318.3 ± 2.9 |
| 3 | Cu/Fe, 100 bar, RT, with additives,d purification on a flash column | 7.8 ± 0.3 |
| 4 | Cu/Fe, 100 bar, RT, with additives,d in line purification with scavenger | 19.4 ± 0.6 |
The development of the Cu/Fe bimetallic catalyst system inspired us to employ iron powder in our CF system as a readily available copper scavenger. Thus, iron powder was filled in a second cartridge which was placed into the flow line according to Fig. 1. We were pleased to find that the utilization of iron powder as scavenger dramatically reduced the copper content of the triazole product (Table 2, entries 2 vs. 4), it proved nearly as effective as a conventional off line purification with column chromatography (entries 3 vs. 4). With in-line copper scavenging, the corresponding 1,4-disubstituted 1,2,3-triazole product was achieved without work-up and purification steps as conversion was quantitative and any excess alkyne could evaporated.
We next investigated the large-scale synthetic capabilities of the Cu/Fe + Fe catalyst and scavenger system with the test reaction between benzyl azide and phenylacetylene at a flow rate of 0.5 mL min−1, a pressure of 100 bar, and at ambient temperature with the joint use of DIEA and AcOH as additives. The concentration and ratio of the starting materials were not changed upon scaling-up. Fractions of the reaction product were collected every 15 minutes, and conversion was determined for each portion. We were pleased to find that the activity of the catalyst remained constant after 150 min of continuous use, and 1.38 g of triazole 1 was obtained without the need for further work-up or purification steps (Table 3). In this experiment, an overall yield of 94% and a productivity of 3 (in mmol product × mmol per catalyst per h) was achieved. We presumed that the iron powder used as scavenger in the large-scale experiment may have catalytic activity in CuAAC reactions due to various copper species adsorbed on its surface. Thus the cartridge containing iron powder after 150 min of continuous copper scavenging was recycled as an in line-generated catalyst column (marked as ‘Cu’/Fe from here on) in a large-scale run utilizing the same reactants and conditions as in the previous experiment (Table 4). It was found that ‘Cu’/Fe effectively catalyzed the CF CuAAC reaction with constant activity in the first 100 min. After that, the activity started to decrease, and in the final fraction a conversion of 31% was obtained. The experiment allowed for the isolation of 1.10 g of pure triazole 1 with a good overall yield of 75%. The SEM images of the iron powder nicely supported the above findings. As shown in Fig. 3a, the surface of the unused iron powder is smooth, but after 150 min of continuous use as a scavenger, small particles appeared on it (Fig. 3b), and the morphology of the surface became similar to that of the Cu/Fe catalyst (Fig. 3b vs. 2). The EDX measurements verified our suspicion that the small dots on the surface of ‘Cu’/Fe are copper nanoparticles, which allowed for catalytic activity in the CuAAC reaction. Thus, the above sustainable procedure allows not only for CF triazole formation with in line copper scavenging but, at the same time, its efficacy for continuous catalyst generation is also noted. The fact that the iron powder used as scavenger in continuo for 150 min works nicely as an in situ generated ‘Cu’/Fe catalyst and the SEM experiments lead us to suggest that, under the present conditions, the scavenger cartridge should be replaced every 2.5 h to maintain the maximum copper scavenging efficiency. However, the use of a larger amount of iron powder (e.g. in a longer column) would possibly allow a longer period with constant efficacy.
|
|||||
|---|---|---|---|---|---|
| Fractiona | Time (min) | Conversion of 1b (%) | Fractiona | Time (min) | Conversion of 1b (%) |
| a 1.38 g of 1 was collected in the whole experiment. An overall yield of 94% and a productivity of 3 (in mmol product × mmol per catalyst per h) was obtained.b Determined by 1H NMR spectroscopic analysis of the crude material. | |||||
| 1 | 0–15 | Quant. | 6 | 75–90 | Quant. |
| 2 | 15–30 | Quant. | 7 | 90–105 | Quant. |
| 3 | 30–45 | Quant. | 8 | 105–120 | Quant. |
| 4 | 45–60 | Quant. | 9 | 120–135 | Quant. |
| 5 | 60–75 | Quant. | 10 | 135–150 | Quant. |

|
|||||
|---|---|---|---|---|---|
| Fractiona | Time (min) | Conversion of 1b (%) | Fractiona | Time (min) | Conversion of 1b (%) |
| a 1.10 g of 1 was collected in the whole experiment. An overall yield of 75% was obtained.b Determined by 1H NMR spectroscopic analysis of the crude material. | |||||
| 1 | 0–15 | Quant. | 6 | 75–90 | Quant. |
| 2 | 15–30 | Quant. | 7 | 90–105 | 99 |
| 3 | 30–45 | Quant. | 8 | 105–120 | 56 |
| 4 | 45–60 | Quant. | 9 | 120–135 | 37 |
| 5 | 60–75 | Quant. | 10 | 135–150 | 31 |

 | ||
| Fig. 3 SEM images of the iron powder: (a) unused iron powder; (b) used iron powder, sample taken after 150 min of CF operation as scavenger (see Table 3). | ||
To investigate the applicability of the CF method, a wide array of CuAAC reactions were carried out utilizing the Cu/Fe catalytic system at a flow rate of 0.5 mL min−1, a pressure of 100 bar, with the joint use of DIEA and AcOH as additives at ambient temperature. At first, the azide scope was examined, employing phenylacetylene as dipolarophile. As can be seen in Table 5, excellent results were achieved with either aliphatic or aromatic azides. In the case of aromatic azides, it was found that either electron-withdrawing (entries 2–4) or electron-donating substituents (entries 5 and 6) are nicely tolerated on the aromatic ring. High conversions and yields were achieved with cyclic, linear, branched and unsaturated non-aromatic azides as well (entries 7–12). Reactions of a series of alkynes with benzyl azide were also carried out. It was found that, besides phenylacetylene, non-aromatic alkynes, such as pent-1-yne, ethyl propiolate and propargyl acetate, are nicely tolerated (entries 13–16). Ferrocene–triazole conjugates are widely applied in medicinal chemistry for the labeling and detection of various systems.25 Thus, CuAAC of benzyl azide with ethynyl ferrocene was also attempted, and ferrocene-substituted triazole 18 was obtained in an excellent yield of 90% (entry 18). In the above experiments the corresponding 1,4-disubstituted 1,2,3-triazole isomers were formed regioselectively, and formation of the 1,5-disubsituted products was not observed. No work-up or purification steps were necessary when the triazole formation was quantitative and any excess alkyne could be volatilized on evaporation (entries 1–6 and 8–12).
| Entry | Azide (1 equiv.) | Alkyne (1.5 equiv.) | Product | Conv.b (%) | Yieldc (%) |
|---|---|---|---|---|---|
| a Conditions: cazide = 0.085 M, 100 bar, RT, 0.5 mL min−1 flow rate, with 0.04 equiv. DIEA + 0.04 equiv. AcOH.b Determined by 1H NMR spectroscopic analysis of the crude material.c Yield of isolated product. | |||||
| 1 |  |
 |
 |
Quant. | 98 |
| 2 |  |
 |
 |
99 | 97 |
| 3 |  |
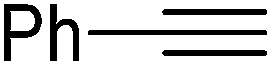 |
 |
Quant. | 98 |
| 4 | 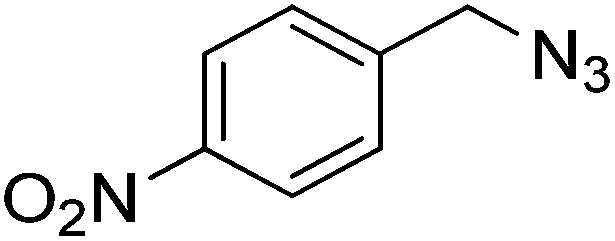 |
 |
 |
Quant. | 96 |
| 5 |  |
 |
 |
Quant. | 98 |
| 6 |  |
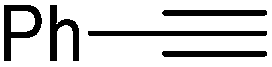 |
 |
99 | 94 |
| 7 |  |
 |
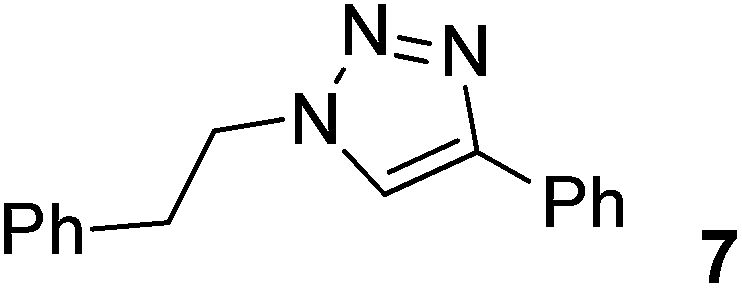 |
97 | 93 |
| 8 |  |
 |
 |
Quant. | 98 |
| 9 |  |
 |
 |
Quant. | 98 |
| 10 | 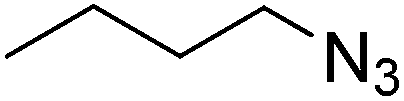 |
 |
 |
Quant. | 93 |
| 11 |  |
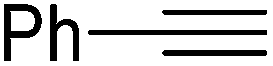 |
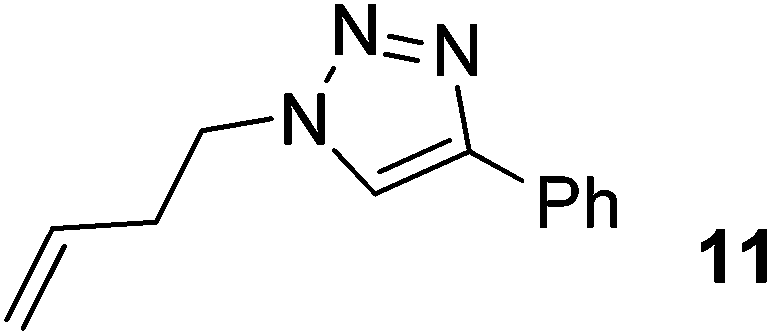 |
Quant. | 98 |
| 12 |  |
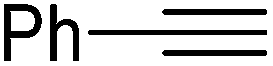 |
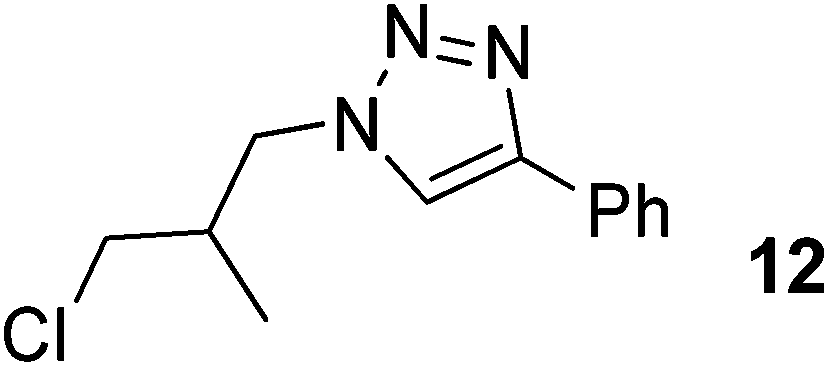 |
Quant. | 97 |
| 13 |  |
 |
 |
90 | 83 |
| 14 |  |
 |
 |
Quant. | 98 |
| 15 |  |
 |
 |
94 | 86 |
| 16 |  |
 |
 |
83 | 75 |
| 17 |  |
 |
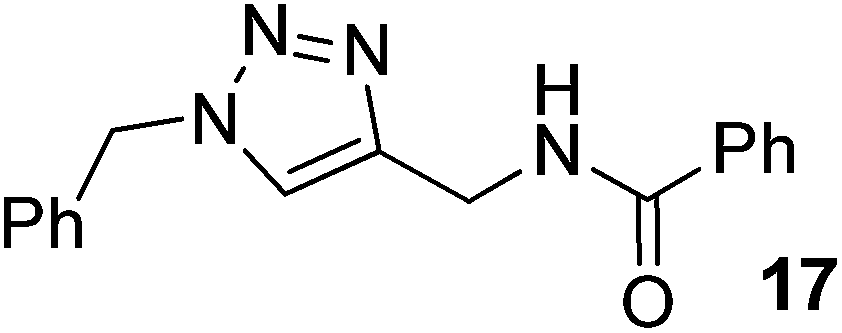 |
Quant. | 92 |
| 18 | 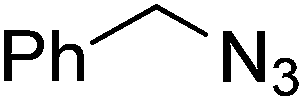 |
 |
 |
Quant. | 90 |
| 19 | 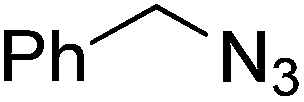 |
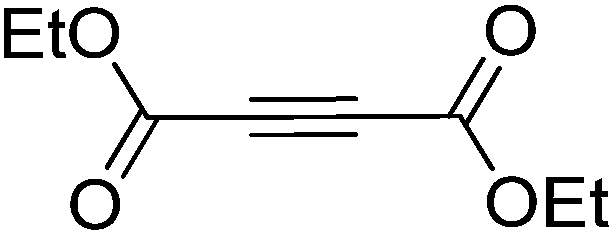 |
 |
Quant. | 90 |
| 20 |  |
 |
 |
27 | 21 |
| 21 |  |
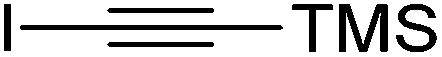 |
 |
55 | 43 |
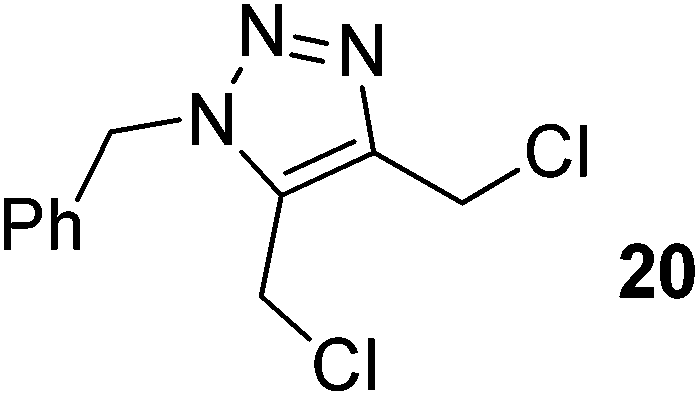
Cycloaddition of disubstituted acetylenes with azides leads to synthetically useful 1,4,5-trisubstituted 1,2,3-triazoles, however the scope of this transformation is mostly limited to 1-haloalkynes.26 Other internal acetylenes have only rarely been used as dipolarophiles in CuAAC reactions,27 mainly due to the fact that the activation toward cycloaddition via coordination of Cu(I) to the alkyne without the possibility of deprotonation involves a significantly higher energetic barrier than is the case with terminal alkynes.28 However, the high activity of the Cu/Fe catalyst system in CuAAC prompted us to test its reactivity with internal alkynes as well. Reactions of benzyl azide with various disubstituted acetylenes were thus explored. We found that in case of electron-deficient alkynes the use of the CF system with the Cu/Fe catalyst (further conditions are as follows: a flow rate of 0.5 mL min−1, a pressure of 100 bar, with 0.04 equiv. DIEA + 0.04 equiv. AcOH at ambient temperature) allowed for the desired trisubstituted triazoles. For example, triazole 19, which is itself a potent antitubercular agent,29 could be obtained from diethyl acetylenedicarboxylate and benzyl azide in quantitative conversion and with a yield of 90% (entry 19). Conversion decreased significantly when more electron rich alkynes, such as 1,4-dichlorobut-2-yne and 1-iodo-2-(trimethylsilyl)acetylene, were reacted (entries 20 and 21). Formation of trisubstituted triazoles was not observed from internal alkynes decorated with two electron donating substituents, such as oct-4-yne and diphenylacetylene. This suggests that the electron density of the triple bond is a crucial factor in the reactions of disubstituted acetylenes.
We would like to emphasize that the investigation of the scope and applicability (Table 5) was fulfilled with a single portion of catalyst without the need for reloading. After this series of experiments no decrease in activity was found. Besides the successful gram-scale synthetic experiment shown in Table 3, this clearly indicates that we managed to create a highly robust system for triazole synthesis.
Conclusions
The efficacy of Cu/Fe bimetallic catalyst system for CuAAC reactions was evaluated in a continuously operating system. It was found that Cu/Fe acts as an outstandingly active and robust catalyst. Due to the high flow rate and short process time utilized for the cycloaddition, the CF technique proved much more rapid than the corresponding batch method. As an additional advantage, the continuous operation provided a contained environment free from harsh reaction conditions to minimize the hazards associated with the handling of explosive azides. It was shown that a column filled with simple iron powder can act as a readily available copper scavenging unit and decrease the copper contaminations of the triazole products significantly. With the opportunity of in-line copper scavenging, the level of copper concentrations can readily be reduced to negligibly small amounts and synthetically useful 1,2,3-triazoles can be obtained in a simple and easy manner without any purification or work-up steps. After several hours of continuous use as scavenger, the iron powder can be reused as an in situ formed catalyst for CuAAC reactions thereby extending the synthetic capabilities of the methodology. The scope and applicability of the CF process was found extremely wide for different azides and alkynes. Not only terminal alkynes, but electron deficient disubstituted acetylenes were also tolerated as dipolarophiles, leading to synthetically useful 1,4,5-trisubstituted 1,2,3-triazoles.Acknowledgements
We are grateful to the Hungarian Research Foundation (OTKA NK81371 and PD103994) and TÁMOP-4.2.2/A-11/1/KONV-2012-0035 for financial support. S. B. Ötvös acknowledges TÁMOP 4.2.4.A/2-11-1-2012-0001 ‘National Excellence Program’, the project was supported by the European Union and the State of Hungary, co-financed by the European Social Fund. I.M.M. acknowledges the award of a János Bolyai scholarship from the Hungarian Academy of Sciences. Z.N. acknowledges for the generous support of “Lendület” Research Scholarship Fund of the Hungarian Academy of Sciences (LP2012-48/2012), and OTKA-NKTH (CK80763). The authors also thank Prof. Tim Peelen (Lebanon Valley College) for the proofreading of this manuscript.References
- C.-K. Sha and A. K. Mohanakrishnan, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, 2003, pp. 623–679 Search PubMed.
- R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- (a) K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC; (b) X. Li, Chem.–Asian J., 2011, 6, 2606 CrossRef CAS; (c) D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399 CrossRef CAS; (d) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC.
- (a) S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696 CrossRef CAS PubMed; (b) A. D. Moorhouse and J. E. Moses, ChemMedChem, 2008, 3, 715 CrossRef CAS PubMed.
- T. Wirth, Microreactors in Organic Chemistry and Catalysis, John Wiley & Sons, 2013 Search PubMed.
- (a) C. Wiles and P. Watts, Green Chem., 2014, 16, 55 RSC; (b) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384 CrossRef CAS PubMed; (c) C. Wiles and P. Watts, Green Chem., 2012, 14, 38 RSC; (d) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17 CrossRef CAS; (e) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS PubMed.
- (a) V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746 CrossRef CAS PubMed; (b) J.-i. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC.
- (a) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC; (b) S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed; (c) J.-i. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed.
- (a) N. Kockmann, M. Gottsponer and D. M. Roberge, Chem. Eng. J., 2011, 167, 718 CrossRef CAS PubMed; (b) C. J. Smith, N. Nikbin, S. V. Ley, H. Lange and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1938 RSC; (c) P. J. Nieuwland, R. Segers, K. Koch, J. C. M. van Hest and F. P. J. T. Rutjes, Org. Process Res. Dev., 2011, 15, 783 CrossRef CAS.
- (a) B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235 CrossRef CAS PubMed; (b) M. Fuchs, W. Goessler, C. Pilger and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 323 CrossRef CAS.
- (a) C. Girard, E. Önen, M. Aufort, S. Beauvière, E. Samson and J. Herscovici, Org. Lett., 2006, 8, 1689 CrossRef CAS PubMed; (b) C. D. Smith, I. R. Baxendale, S. Lanners, J. J. Hayward, S. C. Smith and S. V. Ley, Org. Biomol. Chem., 2007, 5, 1559 RSC; (c) E. Ozkal, S. Ozcubukcu, C. Jimeno and M. A. Pericas, Catal. Sci. Technol., 2012, 2, 195 RSC; (d) E. Ozkal, P. Llanes, F. Bravo, A. Ferrali and M. A. Pericas, Adv. Synth. Catal., 2014, 356, 857 CrossRef CAS.
- (a) S. B. Ötvös, I. M. Mándity, L. Kiss and F. Fülöp, Chem.–Asian J., 2013, 8, 800 CrossRef PubMed; (b) S. B. Ötvös, Á. Georgiádes, I. M. Mándity, L. Kiss and F. Fülöp, Beilstein J. Org. Chem., 2013, 9, 1508 CrossRef PubMed.
- (a) A. R. Bogdan and N. W. Sach, Adv. Synth. Catal., 2009, 351, 849 CrossRef CAS; (b) A. R. Bogdan and K. James, Chem.–Eur. J., 2010, 16, 14506 CrossRef CAS PubMed; (c) S. Ceylan, T. Klande, C. Vogt, C. Friese and A. Kirschning, Synlett, 2010, 2009 CAS; (d) A. R. Bogdan and K. James, Org. Lett., 2011, 13, 4060 CrossRef CAS PubMed.
- A. C. Varas, T. Noël, Q. Wang and V. Hessel, ChemSusChem, 2012, 5, 1703 CrossRef CAS PubMed.
- (a) S. Kovacs and Z. Novak, Org. Biomol. Chem., 2011, 9, 711 RSC; (b) S. Kovács, K. Zih-Perényi, Á. Révész and Z. Novák, Synthesis, 2012, 44, 3722 CrossRef PubMed; (c) A. Székely, Á. Sinai, E. Tóth and Z. Novák, Synthesis, 2014, 46, 1871 CrossRef PubMed.
- Z. Gonda and Z. Novak, Dalton Trans., 2010, 39, 726 RSC.
- R. Hudson, C.-J. Li and A. Moores, Green Chem., 2012, 14, 622 RSC.
- (a) V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210 CrossRef CAS PubMed; (b) C. Shao, X. Wang, Q. Zhang, S. Luo, J. Zhao and Y. Hu, J. Org. Chem., 2011, 76, 6832 CrossRef CAS PubMed.
- (a) C. Nolte, P. Mayer and B. F. Straub, Angew. Chem., Int. Ed., 2007, 46, 2101 CrossRef CAS PubMed; (b) C. Shao, G. Cheng, D. Su, J. Xu, X. Wang and Y. Hu, Adv. Synth. Catal., 2010, 352, 1587 CrossRef CAS; (c) C. Shao, X. Wang, J. Xu, J. Zhao, Q. Zhang and Y. Hu, J. Org. Chem., 2010, 75, 7002 CrossRef CAS PubMed.
- N. Tu, J. Hochlowski and S. Djuric, Mol. Diversity, 2012, 16, 53 CrossRef CAS PubMed.
- V. Ganesh, V. S. Sudhir, T. Kundu and S. Chandrasekaran, Chem.–Asian J., 2011, 6, 2670 CrossRef CAS PubMed.
- (a) B. H. M. Kuijpers, G. C. T. Dijkmans, S. Groothuys, P. J. L. M. Quaedflieg, R. H. Blaauw, F. L. van Delft and F. P. J. T. Rutjes, Synlett, 2005, 3059 CAS; (b) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS PubMed; (c) J. Garcia-Alvarez, J. Diez and J. Gimeno, Green Chem., 2010, 12, 2127 RSC; (d) M. Juríček, K. Stout, P. H. J. Kouwer and A. E. Rowan, Org. Lett., 2011, 13, 3494 CrossRef PubMed.
- (a) S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558 CrossRef PubMed; (b) F. Cuevas, A. I. Oliva and M. A. Pericas, Synlett, 2010, 1873 CAS; (c) R. K. Arigela, A. K. Mandadapu, S. K. Sharma, B. Kumar and B. Kundu, Org. Lett., 2012, 14, 1804 CrossRef CAS PubMed.
- R. Berg and B. F. Straub, Beilstein J. Org. Chem., 2013, 9, 2715 CrossRef CAS PubMed.
- P. Shanmugavelan, S. Nagarajan, M. Sathishkumar, A. Ponnuswamy, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2011, 21, 7273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07954j |
| This journal is © The Royal Society of Chemistry 2014 |