
Microwave-assisted derivatization for fast and efficient analysis of saccharides on disposable microchips†
Abstract
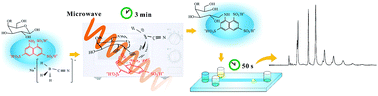
For rapid and low-cost determination of saccharides with microchip electrophoresis, a domestic microwave oven was used to perform the derivatization of saccharides with 8-aminonaphthalene-1,3,6-trisulfonate (ANTS) for laser-induced fluorescence detection with a 405 nm laser diode. The influence of the reaction conditions was systematically examined. Derivatization of xylose, glucose and lactose was fulfilled within 3 min under the optimized condition. With appropriate selection of the separation medium, these model analytes could be resolved within 30 s in borate solutions containing hydroxypropyl cellulose using disposable cyclic olefin copolymer microchips. Theoretical plate numbers of 1.0 × 106 m−1 and limits of detection of 0.15–0.23 μM were achieved. Peak areas were linear up to 1000 μM for all analytes. The applicability of the proposed method was verified by the determination of glucose in human urine and serum. Separation of oligosaccharides from konjac powder was also demonstrated. The domestic microwave oven, the cheap laser diode and disposable microchips used here make the method an economical way for rapid analysis of saccharides.
 Please wait while we load your content...
Please wait while we load your content...

