
Paper-based molecular diagnostic for Chlamydia trachomatis†
Abstract
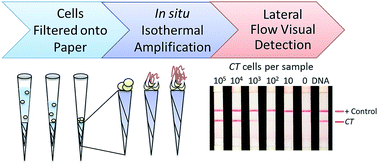
Herein we show the development of a minimally instrumented paper-based molecular diagnostic for point of care detection of sexually transmitted infections caused by Chlamydia trachomatis. This new diagnostic platform incorporates cell lysis, isothermal nucleic acid amplification, and lateral flow visual detection using only pressure and heat sources, eliminating the need for expensive laboratory equipment. This paper-based test can be performed in less than one hour and has a clinically relevant limit of detection that is 100× more sensitive than current rapid immunoassays used for chlamydia diagnosis.
 Please wait while we load your content...
Please wait while we load your content...

