DOI:
10.1039/C4RA07908F
(Paper)
RSC Adv., 2014,
4, 51374-51380
Investigation of the regeneration kinetics of organic dyes with pyridine ring anchoring groups by scanning electrochemical microscopy†
Received
31st July 2014
, Accepted 3rd October 2014
First published on 3rd October 2014
Abstract
This work reports on a study of regeneration kinetics of organic dyes with pyridine ring group sensitized NiO electrodes in combination with iodide-based and organic thiolate-based electrolytes by scanning electrochemical microscopy (SCEM) with a feedback model. These dyes have showed promising performance in p-type dye-sensitized solar cells. The investigation reveals that the anchoring group affects the effective rate constant, showing an efficient dye-regeneration process for the dye with carboxylic acid anchoring groups. Meanwhile, it is worth noting that the regeneration process between the reduced dye and the oxidized state of the thiolate-based electrolyte is much faster than that of the iodide-based electrolyte.
Introduction
The worldwide demand for energy is expected to double by the next decade. An abundant supply of energy is necessary for global political, economic and environmental stability.1–4 Recently, third generation photovoltaic technology, including nanocrystal solar cells and organic solar cells, has attracted much attention due to low cost, simple fabrication relative to others.5,6 Mesoscopic solar cells offer credible and attractive alternative to solid-state p–n junction devices, which efficient transfer solar energy to storable energy based on photosynthesis. For example, dye-sensitized solar cells (DSSC) are under intensive investigation, the promising solution to future large-scale energy conversion issues. Conventional DSSCs typically contain several components such as mesoporous semiconductor metal oxide film, sensitizer, electrolyte/hole conductor, and counter electrode.7,8 An efficient DSSC device depends on several coupled processes.2,9 Upon excitation, the sensitizer injects electron and hole into the n- and the p-type selective contacts, respectively, resulting in the photon-generated charge carriers in femtosecond time regime, which are collected at the front and back electrodes. The photovoltage developed by the cell corresponds to the difference in the quasi-Fermi levels of the n- and the p-type selective contact materials under illumination. It is significant that the positively charged oxidized/photo excited dye D+ is restored to ground state D in nanosecond time regime by receiving an electron from the reduced specie electrolyte (usually being I−).9,10 Mesoporous solar cell configuration has great advantage because this solar energy conversion process involves only majority charge carriers.
To date, an optimum light to electrical energy conversion efficiency of 11.5% has been obtained for ruthenium complex-based DSSCs under reporting conditions with a liquid-junction device configuration.11–13 Even higher efficiencies of 12.3–13% have recently been reported for DSSCs with porphyrin sensitizers and cobalt-based mediators.3,14 Therefore, it is critical to develop new dyes with properties of increased optical cross-section and capable of absorbing longer wavelengths. Other current research focuses on factors including nanomaterials and quantum dots aiming to boost the conversion efficiency. Recently, new type of DSSCs based on sensitization of p-type semiconductor (p-SC, typically NiO) have emerged with great interests.15–18 Contrary to the conventional n-type DSSC, in p-type DSSC the photo-excited sensitizer is reductively quenched by hole injection into the valence band of p-SC.18,19
The efficiency of p-DSSC is intrinsically limited by low open circuit voltage (VOC) and photocurrent. A low photovoltage could be caused by the small difference in potential between the quasi-Fermi level of p-SCs and the electrochemical equilibrium potential of redox mediators.24 The low photocurrent could be related to fast charge recombination between the reduced dye and the holes generated in the NiO, as well as a low light-harvesting efficiency resulting from the limited dye loading on p-type SC film, which leads to low efficiencies.20–23 The role of mediator is continuously to regenerate the reduced sensitizers after hole injection to p-SC valence band and transport charges to the counter electrode. To promote regeneration, redox mediator should satisfy key properties, such fast electron self-exchange rate, and high diffusion coefficient to ensure fast transport of charges to counter electrodes.23,24
Dye regeneration is critical reaction in p-DSSC device, which must be faster than recombination process.25–27 The regeneration kinetics of DSSC depends on various factors, including electrode materials, dye structure, electrolyte composition, and solvent viscosity, etc.11,13,28 Currently, there are few techniques available to study dye-regeneration kinetics at the dye-sensitized nanocrystalline/electrolyte interface in DSSC devices. These are transient adsorption spectroscopy and pump-probe spectroscopy fast incident irradiation.29,30 Scanning electrochemical microscopy (SECM) has been widely used as an effective technique to determining electron transfer kinetics at various interfaces, including polymer/liquid,31 liquid/liquid interfaces,32 redox enzymes on solid supports,33 and SC/electrolyte interfaces.34,35 Recently, Wittstock et al. successfully proposed using SECM to describe the kinetics of dye-regeneration in DSSCs.36–38
In this work, we report on SECM investigation of the influence of anchoring groups on dye-regeneration kinetics at dye-sensitized p-type NiO electrode/electrolyte interface. Specifically, organic sensitizers (P1, CW1, and CW2, see Fig. 1),39 in conjunction with thiolate- and iodide-based electrolytes were selected in order to compare the regeneration process. The regeneration kinetics has been found to depend on the structure of the dye and the concentration of the redox couples, which affect the driving force for regeneration. This contributes to identify of appropriate electrolytes and design of effective sensitizers for highly efficient p-DSSCs.40–42
 |
| | Fig. 1 The structure of dyes (CW1, CW2, and P1) and redox shuttles (thiolate-based and iodide-based electrolytes). | |
Experimental section
Chemicals
The sensitizer P1, CW1, CW2, and redox shuttles (Fig. 1) were synthesized according to literatures.39 Lithium bis(trifluoromethylsulfonyl) imide (LiTFSI) was used as supporting electrolyte. LiI solution was prepared by mixing in 1 mM solution of LiI in acetonitrile with 0.1 M LiTFSI and diluting the resulting stock solution to the required concentration.
Preparation of NiO/dye film
The NiO nanoparticles paste was prepared as fellow 3 mg NiO of nanoparticles (particle size ∼ 20 nm) were ball-milled in 80 mL ethanol overnight. Two kinds of ethyl cellulose (EC) i.e., EC (#5–15 mPa) and EC (#30–60 mPa) were used as binder. The mixture of NiO, EC, triponal, and ethanol was sonicated and stirred over night to obtain fine dispersion.43–45 A paste was made by evaporating ethanol from the mixture with a rotary evaporator. FTO glass were coated with nickel acetate ethanol solution (0.05 M) by dip coating and subsequently dried in air. The photocathode films were screen-printed with the NiO paste and dried for 5 min at 125 °C. This screen printing procedure was repeated to obtain films with different thickness.43–45
Instruments and procedures
SECM feedback mode experiments were performed on CHI 920C electrochemical workstation (CH Instruments, Shanghai). A homemade Teflon cell (with volume of 2 mL) was used to hold a Pt wire counter electrode, an Ag/Ag+ reference electrode. The photocathodes FTO/NiO/organic dyes sample films were attached to the cell bottom and sealed with an O-ring as working electrode. In SECM measurements, the dye-sensitized transparent SC nanocrystal films (NiO with a thickness of about 2.3 μm) were used as the working electrode. An extra Pt wire was connected to FTO substrate with the electrolyte in order to operate the photoelectrochemical cell in a short-circuit setup. A12.5 μm radius Pt wire (Goodfellow, Cambridge, UK) was sealed into a 5 cm glass capillary prepared by a vertical pull pin instrument (PC-10, Japan). The ultra-microelectrode (UME) was polished by a grinding instrument (EG-400, Japan) and micro-polishing cloth with 0.3 μm alumina powder. Then the UME was sharpened conically to a RG of 10, where RG is the ratio between the diameters of the glass sheath and the Pt disk. All experiments were carried out at room temperature. The irradiation was focused onto the backside of the photoanode from light emitting diode.36,37
Result and discussion
In this study, SECM with feedback model was used to analyse the effect of anchoring group on dye regeneration kinetics with various electrolytes under illumination. Two organic dyes with pyridine ring anchoring groups (CW1 and CW2) were synthesized as analogues of P1 dye to better understand the influence of the anchoring group on the regeneration parameters of their corresponding p-DSSCs.39 As shown in Fig. 1, the three dyes contain a common triphenylamine electron-donating group, a single thiophene π-bridge and a 2,2-dicyano-vinyl electron-acceptor group. The CW1 and CW2 dyes contain a pyridine moiety, which acts as a weak electron-withdrawing group and has the same function as the carboxylic acid anchoring group in P1 dye. The introduction of a phenyl conjugate linker between the pyridine ring and the triphenylamine donor produces CW2 with a rigid structure and may stand it up on the NiO surface after dye adsorption, resulting in sufficient dye loading. The device with P1 dye showed photovoltaic efficiency of 0.143% with VOC of 92.9 mV, Jsc of 4.67 mA cm−2 and FF of 0.33. The device with dye CW1, which contains a pyridine moiety as the anchoring group, exhibited VOC of 99 mV, Jsc of 2.66 mA cm−2, and FF of 0.35, thus giving an overall PCE of 0.093%. The device with dye CW2, which contains a phenyl pyridine moiety as the anchoring group, exhibited VOCc of 118 mV, Jsc of 4.05 mA cm−2, and FF of 0.34, giving an overall IPCE of 0.16%.39
Fig. S1 in the ESI† presents the cyclic voltammetry of Pt UME in T− and I− mediator in acetonitrile, corresponding to the steady-state anodic waves according to eqn (1). This measurement was performed in an electrochemical cell with P1 dye-sensitized NiO substrate. Therefore, the reaction on the UME couples with those on the substrate, which are expressed in eqn (2).36 It is evident that the steady state current for I− oxidation varies dramatically compared to T− with the same solvent and concentration. The tip currents for T−-based electrolyte exceeds four times more than I−-based electrolyte.
| | |
2D−/NiO + I−3 → 2D/NiO + 3I−
| (2a) |
| | |
2D−/NiO + T2 → 2D/NiO + 2T−
| (2b) |
In SECM feedback mode under short-circuit condition, the oxidized species of electrolytes (I3− and T2 in this study) are generated by an electrochemical reaction at UME (eqn (1)) and diffuse to dye-sensitized NiO electrode substrates. When the substrate is illuminated from the back, the reduced state of dye molecules (D−) are regenerated by the oxidized species of redox shuttles according to eqn (2), producing the reduced states of redox electrolytes, which eventually diffuse to the UME surface. This process leads to a full cycle reaction between the UME and dye-sensitized substrate. The SECM measurement records the current variations at the UME as a function of normalized distance L (L = d/rT, d being the distance between the tip and the substrate, rT being the radius of the Pt tip) between the UME and the surface of dye-sensitized NiO sample.
Generally, the normalized approach curve in the SECM feedback mode can be described by eqn (3).
| |
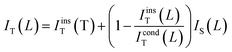 | (3) |
where
IcondT is the current for diffusion-controlled mediator recycling at the sample (positive feedback) and
IinsT describes the current if no reaction occurs at the insulating sample (negative feedback), and
IS(L) (
IS(L) =
IS/
IT,∞) describes the current at the substrate electrode normalized by the current of the UME in the mediator solution (see ESI
†).
From the mathematical description of the mass transport and kinetics at the surface of dye sensitized NiO sample, first-order rate constant κ can be extracted. Then, the effective heterogeneous rate constant keff can be evaluated with equation of keff = κD/rT, where D is the diffusion coefficient for the redox couple. The diffusion coefficients can be determined from microelectrode steady-state currents.37 The typical values of diffusion coefficients are D = 1.86 × 10−5 cm2 s−1 for I− electrolyte and D = 1.35 × 10−5 cm2 s−1 for T− electrolyte, respectively. The reduction constant kred for regeneration of excited dye by the oxidized state of redox shuttle in p-type DSSC device can be extracted from eqn (4).36
| |
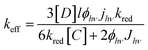 | (4) |
where [
D] is the surface concentration of the dye in the ground state (mol cm
−3),
kred is the heterogeneous rate constant for the reduction of dye by electrolyte, [
C] is the concentration of redox shuttle in electrolyte (mol cm
−3),
l is the film thickness (cm),
ϕhν is the excitation cross-section of the adsorbed dye (cm
2 mol
−1), and
Jhν is the incident photon flux density, respectively.
Fig. 2a shows the normalized experimental approach curves and their corresponding fitting curves according to eqn (3) upon approaching the UME (radius: 12.5 μm) to NiO/P1 film in 0.1 mM I− solution under illumination with a blue LED at different illumination intensities. As the UME approaches the dye-sensitized substrate, the normalized tip current decreases, indicating that tip current depends on the kinetics of reverse reaction on the substrate. Also it was found higher feedback currents for higher light intensities, implying that regeneration rate increases with illumination intensities. When the film was back-illuminated, the current was significantly larger than in the dark (curve insulting surface curve#0). Under illumination photon density of blue LED changed from 2.2 × 109 to 22.4 × 109 mol cm2 s−1, their corresponding rate constants keff increased from 0.134 × 10−3 to 1.056 × 10−3 cm s−1. In similar pattern, Fig. 2b shows when photon density of red LED increased from 4.19 × 109 to 14.684 × 109 mol cm2 s−1, their corresponding rate constants keff increased from 0.0178 × 10−3 to 0.2484 × 10−3 cm s−1.
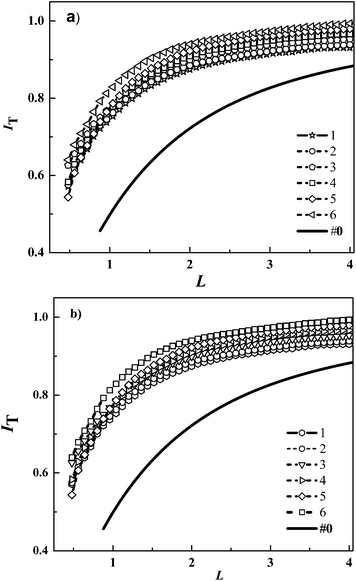 |
| | Fig. 2 (a) Comparison of normalized SECM approach curves for the approach of a Pt disk UME towards a NiO/P1 film in 1 mM I− solution under different illumination. Photon flux density for (a) blue LED (in 10−9 mol cm−2 s−1): (1) 2.2, (2) 6.1, (3)11.8, (4) 13.9, (5) 19.8, (6) 22.4. The first-order rate constant κ was estimated to be: (1) 0.009, (2) 0.013, (3) 0.025, (4) 0.036, (5) 0.05, (6) 0.099, and the effective rate constant keff (10−3 cm s−1) was estimated to be: (1) 0.134, (2) 0.193, (3) 0.372, (4) 0.535, (5) 0.788, (6) 1.473, respectively; and for (b) red LED: (1) 4.19, (2) 6.81, (3) 9.44 (4) 12.06, (5) 13.11, (6) 14.68. The first-order rate constant κ was estimated to be: (1) 0.0012, (2) 0.0021, (3) 0.0048, (4) 0.0076, (5) 0.0116, (6) 0.0167, and the effective rate constant keff: (1) 0.0178, (2) 0.0312, (3) 0.0714, (4) 0.1131, (5) 0.1726, (6) 0.2484. | |
In order to investigate influence of incident light intensity on regeneration kinetics, we performed feedback current distance curve measurement at a fixed concentration of electrolyte. Fig. 3 shows the effective rate constant keff for P1 dye in T− electrolyte and I− electrolyte as a function of photon density, Jhν in different LEDs: (a) for blue LED and (b) for red LED. It was found with a blue LED, the keff for P1 dye in 1 mM T− electrolyte increased from 0.355 × 10−3 to 1.142 × 10−3 cm s−1 as Jhν increased from 2.2 × 10−9 to 22.4 × 10−9 mol cm−2 s−1 (○). The keff for P1 dye in 1 mM I− electrolyte increased from 0.216 × 10−3 to 0.671 × 10−3 cm s−1 (Δ) (Fig. 3a and Table 1). Fig. 3b shows similar observation in keff for P1 dye-sensitized with red LED. The keff increased from 0.319 × 10−3 to 0.941 × 10−3 cm s−1 (Δ) as the Jhν increased from 4.19 × 10−9 to 14.68 × 10−9 mol cm−2 s− in 1 mM I− electrolyte, while they increased from 0.381 × 10−3 to 1.401 × 10−3 cm s−1 (○) in 1 mM T− electrolyte (Fig. 3b and Table 1). This confirms that the effective rate constant keff of P1 dye with T− electrolyte is significantly higher than I− electrolyte.
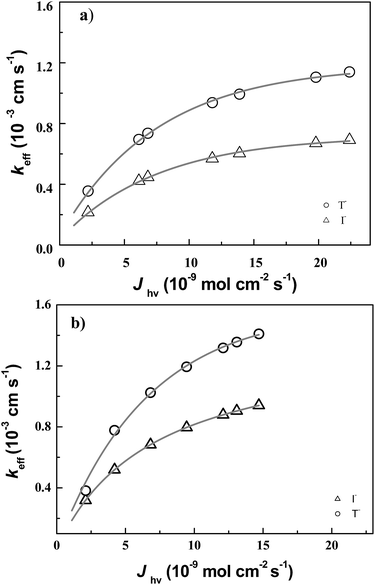 |
| | Fig. 3 Plot of experimental values of keff as a function of Jhν(λ). The lines represent fits of keff according to eqn (3) with ked and Jhν(λ) as adjustable parameters for T− electrolyte (○) and I− electrolyte (Δ) illuminated with (a) blue LED and (b) red LED. | |
Table 1 Normalized apparent heterogeneous first-order rate constants κ and apparent heterogeneous rate constants (keff = κD/rT) obtained for the reduction of photo-excited P1 (rT = 12.5 μm, RG = 10, D(T−) = 1.35 × 10−5 cm2 s−1, and D(I−) = 1.86 × 10−5 cm2 s−1)
| Jhν [10−9 mol cm−2 s−1] |
keff(T−) [10−3 cm s−1] |
keff(I−) [10−3 cm s−1] |
| (a) Blue LED |
| 2.2 |
0.216 |
0.202 |
| 6.1 |
0.695 |
0.422 |
| 11.8 |
0.735 |
0.446 |
| 13.9 |
0.937 |
0.569 |
| 19.8 |
0.993 |
0.604 |
| 22.4 |
1.142 |
0.671 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| (b) Red LED |
| 4.19 |
0.381 |
0.319 |
| 6.81 |
0.777 |
0.519 |
| 9.41 |
1.025 |
0.683 |
| 12.06 |
1.193 |
0.709 |
| 13.11 |
1.317 |
0.904 |
| 14.68 |
1.401 |
0.941 |
Therefore, the reduction constant kred for regeneration of excited dye and the excitation cross-section (ϕhν) of the adsorbed dye was evaluated by using eqn (4), exhibiting significant relationship with electrolytes and irradiation intensity. The calculated reduction constant kred for P1 were 3.53 × 105 and 9.67 × 105 mol−1 cm3 s−1 in I− and T− electrolytes, respectively. The values of ϕhν for P1 dye were calculated to be 9.02 × 10−5 and 1.79 × 10−5 cm2 mol−1 in I− electrolyte illuminated with blue and red LEDs. The values of ϕhν for P1 dye were 1.28 × 10−4 and 1.23 × 10−5 cm2 mol−1 in T− electrolyte illuminated with blue and red LEDs, respectively. This result could be attributed to the remarkable dependence of heterogeneous rate constant of dye regeneration on wavelength and intensity.46,47
Based on above observation, SECM investigation dye-regeneration kinetics of dye with different anchoring group was further carried out in order to understand influence of dye natures. Fig. 4 shows the normalized current distance curves and corresponding fitting data for (a) CW1 dye and (b) CW2 dye sensitised NiO films. Under illumination with blue LED for CW1 sensitized NiO film in 1 mM I−, keff was increased from 0.014 × 10−3 to 0.171 × 10−3 cm s−1 as the photon flux of blue LED from 2.2 × 10−9 to 22.4 × 10−9 mol cm2 s−1. For red LED of photon density increased from 4.19 × 10−9 to 14.68 × 10−9 mol cm2 s−1, the keff increased from 0.265 × 10−3 to 0.782 × 10−3 cm s−1 for CW1. In same pattern for CW2-senstized NiO films, the keff increased from 0.145 × 10−3 to 0.182 × 10−3 cm s−1 with blue LED and keff increased from 0.167 × 10−3 to 0.492 × 10−3 cm s−1 with red LED (see Table 2).
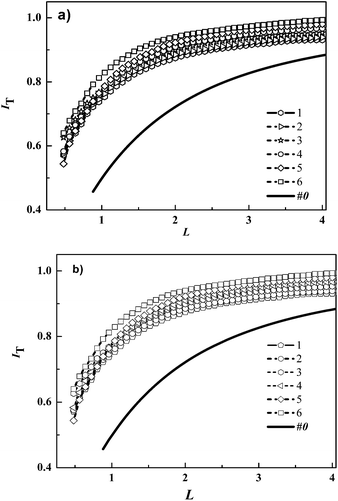 |
| | Fig. 4 (a) Comparison of normalized SECM approach curves between Normalized SECM feedback approach curves for the approach of a Pt disk UME towards a NiO/CW1 and NiO/CW2 film in the dark (curve 1) and under illumination by blue LED. Photon flux density of LED in 10−9 mol cm−2 s−1 (a) NiO/CW2 (1) 2.2, (2) 6.1, (3) 11.8, (4) 13.9, (5) 19.8, (6) 22.4; VT = 0.05 V s−1, ET = 0.7 V, [I−] = 1 mM. keff × 10−3 (1) 0.145, (2) 0.148, (3) 0.155, (4) 0.162, (5) 0.171, (6) 0.182 (b) NiO/CW1, keff × 10−3 (1) 0.014, (2) 0.111, (3) 0.141, (4) 0.149, (5) 0.166, (6) 0.171. | |
Table 2 Normalized apparent heterogeneous first-order rate constants κ and apparent heterogeneous first-order rate constants (keff = κD/rT) obtained for the reduction of photo-excited CW2, CW1, and P1 by I− for different illumination intensity of blue, and red LEDs. [I−] = 1 mM, D = 1.867 × 10−5 cm2 s−1, rT = 12.5 μm, RG = 10
| Jhν [10−9 mol cm−2 s−1] |
CW2 |
CW1 |
P1 |
| keff [10−3 cm s−1] |
keff [10−3 cm s−1] |
keff [10−3 cm s−1] |
| (a) Blue LED |
| 2.2 |
0.145 |
0.014 |
0.202 |
| 6.1 |
0.148 |
0.111 |
0.422 |
| 11.8 |
0.155 |
0.141 |
0.446 |
| 13.9 |
0.162 |
0.149 |
0.569 |
| 19.8 |
0.171 |
0.166 |
0.604 |
| 22.4 |
0.182 |
0.171 |
0.671 |
|
| (b) Red LED |
| 4.19 |
0.167 |
0.265 |
0.319 |
| 6.81 |
0.271 |
0.431 |
0.519 |
| 9.44 |
0.358 |
0.568 |
0.683 |
| 12.06 |
0.417 |
0.662 |
0.709 |
| 13.11 |
0.461 |
0.731 |
0.904 |
| 14.68 |
0.492 |
0.782 |
0.941 |
The influence of light intensities on dye regeneration kinetics was compared for CW1, CW2, and P1 dyes (Table 2). The experimental result clearly shows that keff increases with light intensity. Fig. 5 shows the fitting result of keff vs. Jhν varied light intensity of the three dyes in I− electrolyte as a function of photon flux density Jhν with (a) blue and (b) red illumination. It was found that under a given photon flux intensity, the P1 dye had highest keff values among the three dyes. Under illuminated with blue LED, the keff values were slightly smaller than those with red LED. The CW1 and CW2 dyes presented more or less the same regeneration kinetics rate constant keff. It was also observed that the dye with carboxylic acid anchoring group showed faster regeneration process than that of dye with pyridine as anchoring group. This result indicates that the dye regeneration is dependent on dye molecular structure. The values of ϕhν were extracted from the fitting result with eqn (4) to be 4.51 × 10−5 and 1.06 × 10−5 cm2 mol−1 for CW2 dye illuminated with blue and red LEDs. For CW1 dye ϕhν was calculated to be 3.35 × 10−5 and 2.23 × 10−5 cm2 mol−1 for blue and red LEDs (see Table 3).
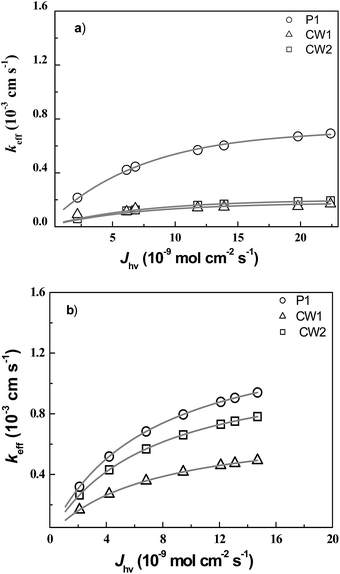 |
| | Fig. 5 Plot of experimental values of keff versus Jhν(λ): (a) for blue and (b) red LEDs. The lines represent fits of keff according to eqn (3) with kred and ϕhν(λ) as adjustable parameters of P1 (○), CW2 (Δ), and CW1 (□) dyes. | |
Table 3 Fitting results of the reduction rate constant kred and cross section ϕhν for three organic dyes from the experimental data of keff, I− electrolyte concentration
| Dye |
LED |
kred [mol−1 cm3 s−1] |
ϕhν [mol−1 cm2] |
| P1 |
Blue |
3.53 × 106 |
4.53 × 10−5 |
| Red |
2.21 × 10−5 |
| CW1 |
Blue |
3.72 × 105 |
1.15 × 10−5 |
| Red |
2.23 × 10−5 |
| CW2 |
Blue |
9.95 × 105 |
3.35 × 10−5 |
| Red |
1.06 × 10−5 |
The SCEM investigation of dye regeneration conforms that change of regeneration parameters k, keff, and kred exhibit significant relationship with incident light intensities. In DSSCs, kinetics of electron transfer reaction at the dye sensitized semiconductor/electrolyte interfaces is critical to device efficiency. Previous report on the P1, CW1 and CW2 dyes revealed their oxidation potential (Eox) at 1.38, 1.179, and 1.109 V (vs. NHE), respectively.39 The redox potential values of I3−/I− and T2/T− were about 0.35 and 0.5 V (vs. NHE). In our experimental observation, the E(D/D−) were estimated to be −3.13 eV for P1 dye, −3.12 eV for CW1, and −3.09 eV for CW2 (Fig. 6). Therefore, the regeneration energy of (ΔGreg) per the elementary charge can be 2.13 eV for P1 dye, 2.3 eV for CW1 dye, and 2.27 eV for CW2.11,27,39 As discussed above, it is significant that the dye regeneration is faster in (T−) electrolyte compared to (I−) electrolyte, even though the driving force is smaller than former Table 4. Therefore, there is a significant driving force for dye regeneration reaction as illustrated in Fig. 6.
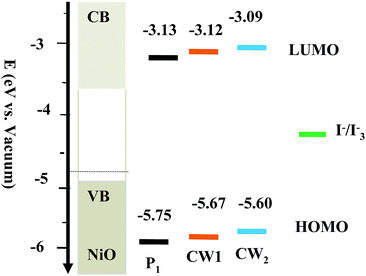 |
| | Fig. 6 Schematic energy level diagram for various dyes with respect to the NiO. | |
Table 4 Fitting results of the reduction rate constant kred and cross section ϕhν for P1 dye in different electrolytes
| Electrolyte |
LED |
kred [mol−1 cm3 s−1] |
ϕhν [mol−1 cm2] |
| I− |
Blue |
3.53 × 106 |
4.53 × 10−5 |
| Red |
2.21 × 10−5 |
| T− |
Blue |
9.67 × 106 |
1.28 × 10−4 |
| Red |
1.23 × 10−5 |
Conclusion
The SECM with feedback mode can provide insight on impact of different parameters on dye regeneration process, including dye molecular structure and redox couples. The experimental results in this study showed that the dye-regeneration rate constant kred for the organic D–π–A dye P1 was about 3.53 × 106 mol−1 cm3 s−1 with an excitation cross section about 4.53 × 10−5 cm2 mol−1 (blue illumination) with the standard I− electrolyte. The dye regeneration process can be boosted by using thiolate-based electrolyte, showing kred of 9.67 × 106 mol−1 cm3 s−1 with an excitation cross section about 1.28 × 10−4 mol−1 cm2. Changing the carboxylic acid anchoring group with pyridine for CW1 and CW2 dyes, the dye regeneration rate constant of kred were determined to be 3.72 × 105 and 9.95 × 105 mol−1 cm3 s−1, the excitation cross section to be 1.15 × 10−5 and 3.35 × 10−5 cm2 mol−1, respectively. The observed difference is related to electrolyte composition, the internal structure of sensitizers, and the intermolecular interaction of dye and electrolyte. The molecular structure dependence of dye-regeneration process can be attributed to the drive force for the charge transfer reaction. Therefore, SECM measurement allows for rather straight forward test of different electrolyte compositions on the same substrate without the need to construct series of complete cells. This method contributes to selection and design of high efficient electrolytes and sensitizers for improvement p-type DSSC.
Acknowledgements
We gratefully acknowledge the 973 Program of China (2014CB643506, 2013CB922104, and 2011CBA00703), NSFC (21103578, 21161160445, and 201173091), and the CME with the Program of New Century Excellent Talents in University (NCET-10-0416). The authors thank the Analytical and Testing Center of Huazhong University of Science and Technology for support.
Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef
 .
. - A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed .
- S. Nakade, Y. Makimoto, W. Kubo, T. Kitamura and Y. Wada, J. Phys. Chem. B, 2005, 109, 3488–3493 CrossRef CAS PubMed .
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed .
- S. Rhee and W. Kwon, J. Chem. Eng., 2011, 7, 1481–1494 Search PubMed .
- T. Hamann, R. Jensen, B. Martinson, H. Ryswyk and J. Hoppy, Energy Environ. Sci., 2008, 1, 66–78 CAS .
- T. Daeneke, A. Mozer, T. Kwon, N. Duffy, A. Holmes, U. Bach and L. Spiccia, Energy Environ. Sci., 2012, 5, 7090–7099 CAS .
- J. Bandara and H. Weerasinghe, Sol. Energy Mater. Sol. Cells, 2005, 85, 385–390 CrossRef CAS PubMed .
- Y. Tachibana, J. Moser, M. Grätzel, D. Klug and J. Durrant, J. Phys. Chem., 1996, 100, 20056–20062 CrossRef CAS .
- S. Haque, Y. Tachibana, D. Klug and J. Durrant, J. Phys. Chem. B, 1998, 102, 1745–1749 CrossRef CAS .
- E. Gibson, L. Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS PubMed .
- M. Nazeeruddin, A. Kay, I. Rodicio, R. Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS .
- P. Wang, S. Zakeeruddin, J. Moser, M. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS PubMed .
- A. Yella, H. Lee, H. Tsao, C. Yi, A. Chandiran, M. Nazeeruddin, E. Diau, C. Yeh, S. Nazeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed .
- X. Xu, B. Zhang, J. Cui, D. Xiong, Y. Shen, W. Chen, L. Sun, Y. Cheng and M. Wang, Nanoscale, 2013, 5, 7963–7969 RSC .
- X. Xu, J. Cui, J. Han, J. Zhang, Y. Zhang, L. Luan, G. Alemu, Z. Wang, Y. Shen, D. Xiong, W. Chen, Z. Wei, S. Yang, B. Hu, Y. Cheng and M. Wang, Sci. Rep., 2014, 4, 3961 Search PubMed .
- Z. Xu, D. Xiong, H. Wang, W. Zhang, X. Zeng, L. Ming, W. Chen, X. Xu, J. Cui, M. Wang, S. Powar, U. Bach and Y. Cheng, J. Mater. Chem. A, 2014, 2, 2968–2976 CAS .
- F. Odobela, Y. Pellegrin, E. Gibsonb, A. Hagfeldtc, A. Smeig and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef PubMed .
- F. Odobel, L. Pleux, Y. Pellegrin and E. Blart, Acc. Chem. Res., 2010, 43, 1063–1107 CrossRef CAS PubMed .
- J. He, H. Lindstrom, A. Hagfeldt and S. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS .
- A. Nattestad, M. Ferguson, R. Kerr, Y. Cheng and U. Bach, Nanotechnology, 2008, 19, 295304–295313 CrossRef PubMed .
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 194, 143–147 CrossRef CAS PubMed .
- C. Fernando, A. Kitagawa, M. Suzuki, K. Takahashi and T. Komura, Sol. Energy Mater. Sol. Cells, 1994, 33, 301–315 CrossRef CAS .
- L. Li, E. Gibson, P. Gibson, G. Qin, M. Boschloo, A. Gorlov, L. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef CAS PubMed .
- X. Wu, G. Xing, L. Tan, Y. Webster, T. Sum and K. Edwin, Phys. Chem. Chem. Phys., 2012, 14, 9511–9519 RSC .
- X. Zhang, F. Huang, A. Nattestad, K. Wang, D. Fu, A. Mishra, P. Bäuerle, U. Bach and Y. Cheng, Chem. Commun., 2011, 47, 4808–4810 RSC .
- P. Qin, M. Linder, T. Brinck, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Mater., 2009, 21, 2993–2996 CrossRef CAS .
- M. Griffith, K. Sunahara, A. Furube, A. Mozer, L. David, P. Wagner, G. Wallace and S. Mori, J. Phys. Chem. C, 2013, 117, 1885–1889 Search PubMed .
- P. Qin, J. Wiberg, E. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CAS .
- S. Ito, T. Murakami, P. Comte, P. Liska, C. Grätzel, M. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS PubMed .
- G. Wittstock, M. Burchardt, S. Pust, Y. Shen and C. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1584–1617 CrossRef CAS PubMed .
- P. Sun Franc, O. Laforge and M. Mirkin, Phys. Chem. Chem. Phys., 2007, 9, 802–823 RSC .
- J. Amphlett and G. Denuault, J. Phys. Chem. B, 1998, 102, 9946–9951 CrossRef CAS .
- C. Wei and A. Bard, J. Phys. Chem., 1995, 99, 16033–16042 CrossRef CAS .
- Y. Shao and M. Mirkin, J. Phys. Chem. B, 1998, 102, 9915–9921 CrossRef CAS .
- Y. Shen, K. Nonomura, D. Schlettwein, C. Zhao and G. Wittstock, Chem.–Eur. J., 2006, 12, 5832–5839 CrossRef CAS PubMed .
- Y. Shen, U. Tefashe, K. Nonomura, T. Lowenstein, D. Schlettwein and G. Wittstock, Electrochim. Acta, 2009, 55, 458–464 CrossRef CAS PubMed .
- B. Zhang, X. Xu, X. Zhang, D. Huang, S. Li, Y. Zhang, F. Zhan, M. Deng, Y. He, W. Chen, Y. Shen and M. Wang, ChemPhysChem, 2014, 15, 1182–1189 CrossRef CAS PubMed .
- J. Cui, J. Lu, X. Xu, K. Cao, Z. Wang, G. Alemu, H. Yuang, Y. Shen, J. Xu, Y. Cheng and M. Wang, J. Phys. Chem. C, 2014, 118, 16433–16440 CAS .
- L. Lu, J. Bai, X. Xu, Z. Li, K. Cao, J. Cui and M. Wang, Chin. Sci. Bull., 2012, 57, 4131–4142 CrossRef .
- M. Wang, C. Grätzel, S. Zakeer and M. Grätzel, Energy Environ. Sci., 2012, 5, 9394–9405 CAS .
- M. Wang, N. Chamberland, L. Breau, J. Moser, R. Humphry-Baker, B. Marsan, S. Zakeeruddin and M. Grätze, Nat. Chem., 2010, 2, 385–389 CrossRef CAS PubMed .
- X. Zhang, F. Huang, A. Nattestad, K. Wang, D. Fu, A. Mishra, P. Bäuerle, U. Bach and Y. Cheng, Chem. Commun., 2011, 47, 4808–4810 RSC .
- X. Zhang, Z. Zhang, D. Chen, P. Bauerle, U. Bach and Y. Cheng, Chem. Commun., 2012, 48, 9885–9887 RSC .
- S. Ito, T. Murakami1, P. Comte, P. Liska, C. Grätzel, M. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS PubMed .
- S. Powar, T. Daeneke, M. Ma, D. Fu, N. Duffy, G. Gçtz, M. Weidelener, P. Bauerle, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 602–605 CrossRef CAS PubMed .
- G. Alemu, B. Zhang, J. Li, X. Xu, J. Cui, Y. Shen and M. Wang, NANO, 2014, 09, 1440008 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07908f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*







