
Scalable asymmetric synthesis of a key fragment of Bcl-2/Bcl-xL inhibitors†
Sylvain Laclef‡
a,
Catherine Taillier‡a,
Christine Penloupb,
Aurélie Vigerb,
Jean-François Brièrea,
Christophe Hardouin*b and
Vincent Levacher*a
aNormandie UNIV, COBRA, UMR 6014 et FR3038; Univ Rouen; INSA Rouen; CNRS, IRCOF, 1 rue Tesnière, 76821 Mont Saint Aignan Cedex, France. E-mail: vincent.levacher@insa-rouen.fr
bOril Industrie, 13 rue Auguste Desgenétais, 76210 Bolbec, France. E-mail: christophe.hardouin@fr.netgrs.com
First published on 8th August 2014
Abstract
The asymmetric synthesis of a 1,3-diamine building block for the elaboration of Bcl-2 and Bcl-xL protein inhibitors is described through two key steps: (1) a highly diastereoselective aza-Reformatsky reaction, and (2) a chemoselective amination under Mitsunobu conditions. This synthetic sequence was also demonstrated to be successfully amenable to a large-scale synthesis.
Defects in the apoptotic processes1 play an important role in tumour initiation, progression and chemoresistance.2 Among the apoptosis regulator Bcl-2 family (B-cell lymphoma 2), the anti-apoptotic Bcl-2 and Bcl-xL proteins were found to be overexpressed in many cancers.3–5 As part of a complex orchestration to regulate cell fate, the anti-apoptotic Bcl-2 and Bcl-xL proteins and others inhibit pro-apoptotic proteins such as BAK and BAX. Importantly, these interactions can be antagonised by BH3-only proteins (BAD, BIM and NOXA) possessing a single BH domain and displaying a large hydrophobic groove with the same fold. Consequently, the development of small molecule BH3 mimetics as inhibitors of anti-apoptotic Bcl-2 and Bcl-xL proteins is attractive for novel anticancer therapy.6–9
A fragment-based drug design10 approach has led to the discovery of several11 potent Bcl-2 and Bcl-xL inhibitors such as 1 in Abbott Laboratories (ABT-737, Fig. 1).12 Analogues based on similar scaffolds were recently developed.13 In that field of research (Fig. 1), Servier Laboratories developed conformationally restricted isosters 2, which displayed submicromolar activity. The tricyclic architecture was aimed at addressing both the solubility issues and at modulating the interactions with the hydrophobic groove of the proteins. Extensive structure–activity relationship studies revealed the essential importance of common diamine fragments such as 3,14 containing a 1,3-diamine moiety flanked by a phenylthioethyl arm, for securing both bioavailability and the potent inhibition of Bcl anti-apoptotic proteins. These outcomes highlighted that the R isomer displayed better bioactivity than the opposite enantiomer.
 | ||
| Fig. 1 Structures of Bcl-2/Bcl-xL inhibitors. | ||
As far as the construction of diamine fragment 3 was concerned, only one chiral pool based synthesis was reported using L-aspartic acid precursor.15 This method allowed the synthesis of compound 3 in eight steps and with a 30% overall yield.
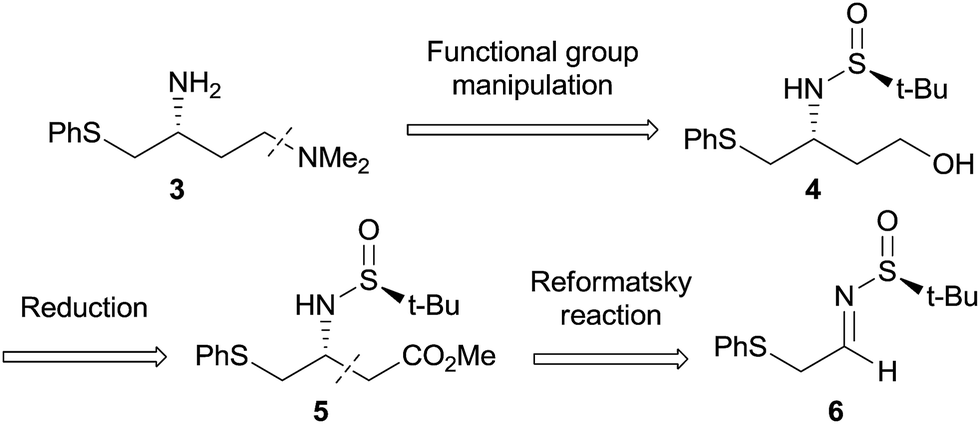
In this context, we endeavoured to develop a reliable access toward diamine 3 through an alternative asymmetric synthesis approach. The aim is eventually to achieve a flexible larger-scale synthetic sequence, en route to providing significant amounts of Bcl-2 protein inhibitor from the key building block 3. The retrosynthetic approach is based on both diastereoselective aza-Reformatsky (6 to 5) and chemoselective amination key reactions (4 to 3, Scheme 1). First, chiral Ellman's N-tert-butanesulfinamide, readily available on a large scale as both enantiomers, was selected as a versatile chiral auxiliary for the asymmetric synthesis of amine 5.16 However, despite previous examples reporting the use of chiral Ellman's sulfinimines in Reformatsky reactions,17 the influence of the thioether functionality of 6 on both reactivity and diastereoselectivity remains an open issue. Then, we sought to capitalize on the N-sulfinyl protecting group of 5, in order to perform further functional group manipulation, like the key chemoselective amination step on 4. We are pleased to report herein our efforts towards the development of a scalable diastereoselective synthesis of chiral scaffold 3, a potentially versatile and useful building block in medicinal chemistry.
 | ||
| Scheme 1 Asymmetric synthesis approach of diamine fragment. | ||
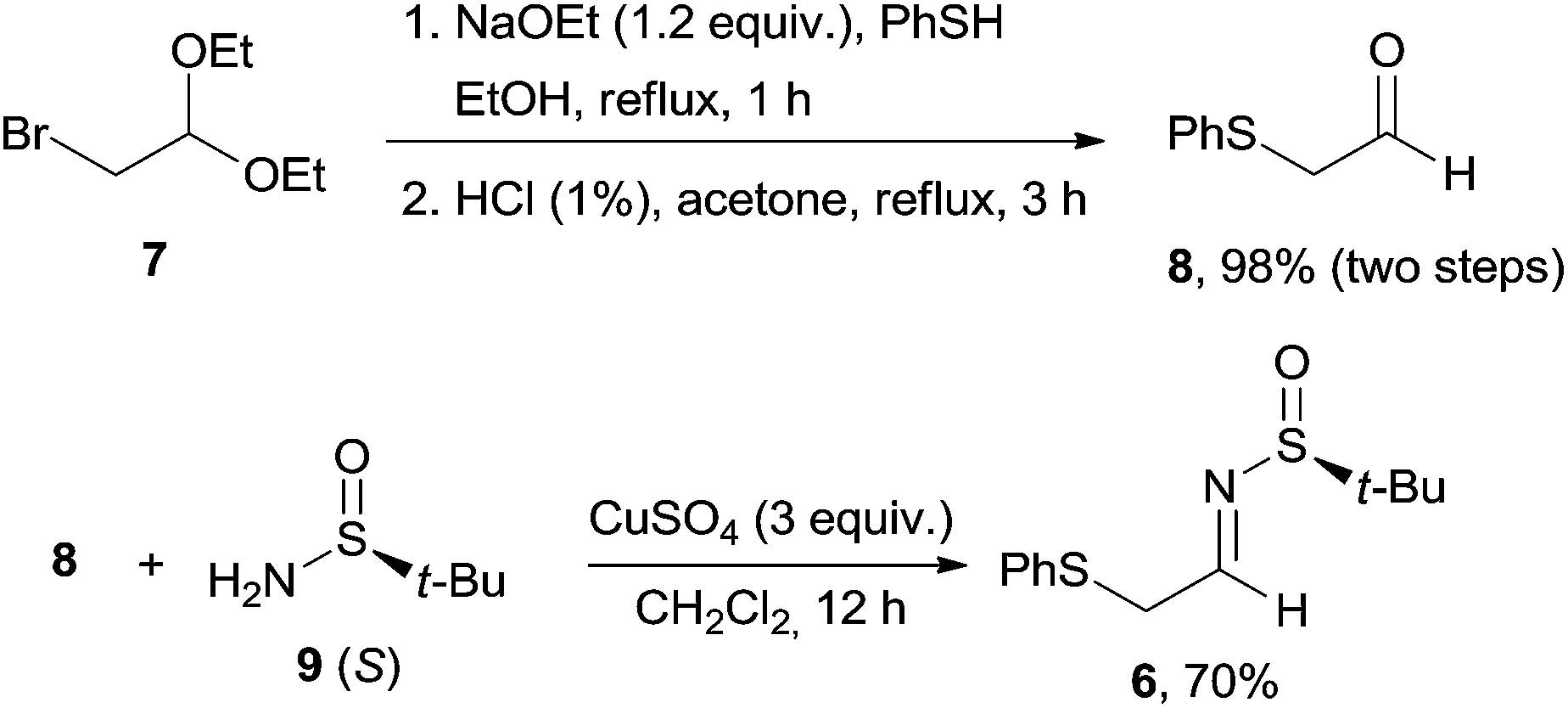
According to literature procedures, bromoacetaldehyde acetal 7 was converted to the aldehyde precursor 8 in two steps (Scheme 2).18 The transformation of the rather unstable aldehyde 8 into the corresponding enantiomerically pure N-(tert-butylsulfinyl)imine 6 was successfully carried out with copper(II) sulfate as the dehydrating agent in a 70% yield.16 These conditions were superior to the standard use of Ti(OEt)4, which gave 6 in only a 52% yield. It should be noted that other chiral auxiliaries, such as (R)-1-phenylethylamine or (R)-2-methoxy-1-phenylethylamine, failed to give the corresponding imines, highlighting the robustness of the Ellman's sulfinamides approach.
 | ||
| Scheme 2 Synthesis of sulfinimine intermediate 6. | ||
Then, the sulfinimine 6 was treated with an excess of the Reformatsky reagent derived from the corresponding bromo acetate 10 (2.2 equiv.) under Barbier's conditions (Scheme 3).19 Pleasingly, the desired product 5 was obtained in a 85% yield, with a high diastereoisomeric ratio (d.r. > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). Actually, changing the reaction temperature from 50 °C to 0 °C had negligible impact on d.r., although it led to lower yields in some cases. By means of Red-Al, the methyl ester 5 was easily reduced, to furnish the desired (R)-alcohol 4 in a 70% yield. Based on Ellman's model, already applied to the Reformatsky reagent originating from 10, we propose the following explanation to account for the diastereoinduction outcome.16,17c Considering that the Reformatsky reagent derived from methyl bromoacetate 10 exists as a monomeric C-metallated species in polar solvents,20 a regular Zimmerman–Traxler transition state involving a six-membered intermediate with zinc metals coordinated to the sulfinyl oxygen is proposed. Then, the nucleophilic attack of the Reformatsky reagent to the Re face of imine takes place (Scheme 3). The high diastereoselectivity obtained demonstrates that the putative coordination between sulfinimine and zinc is not disturbed by other complexing functions such as the thioether moiety.
:5). Actually, changing the reaction temperature from 50 °C to 0 °C had negligible impact on d.r., although it led to lower yields in some cases. By means of Red-Al, the methyl ester 5 was easily reduced, to furnish the desired (R)-alcohol 4 in a 70% yield. Based on Ellman's model, already applied to the Reformatsky reagent originating from 10, we propose the following explanation to account for the diastereoinduction outcome.16,17c Considering that the Reformatsky reagent derived from methyl bromoacetate 10 exists as a monomeric C-metallated species in polar solvents,20 a regular Zimmerman–Traxler transition state involving a six-membered intermediate with zinc metals coordinated to the sulfinyl oxygen is proposed. Then, the nucleophilic attack of the Reformatsky reagent to the Re face of imine takes place (Scheme 3). The high diastereoselectivity obtained demonstrates that the putative coordination between sulfinimine and zinc is not disturbed by other complexing functions such as the thioether moiety.
 | ||
| Scheme 3 The key Reformatsky reaction. | ||
Two different pathways were next considered to transform the primary alcohol 4 into the tertiary amine 11 (Scheme 4). First, following a two-step sequence, the alcohol 4 was converted quantitatively to aldehyde 12 using the mild 2,2,6,6-tetramethyl-1-piperidinyloxyl and [bis(acetoxy)-iodo]benzene (TEMPO-BAIB) oxidative system (Route A, Scheme 4).21 Then, the crude aldehyde 12 underwent a reductive amination sequence in the presence of NaBH(OAc)3 to give amine 11 in a 72% overall yield. Unfortunately, we encountered significant reproducibility issues due to the instability of aldehyde 12 when attempting to scale up the reaction. To overcome these difficulties, an alternative approach (Route B) based on a one-step Mitsunobu reaction with dimethylamine was studied.22 This strategy led to the formation of product 11, with a respectable yield of 43% and, more importantly, with a robust scalable protocol (vide infra). Finally, the deprotection of the N-sulfinyl functional group of 11 was achieved under regular acidic conditions, affording the target diamine molecule 3 in a 87% yield. The R-absolute configuration was assigned at that stage by comparison with the optical rotation previously reported.10
 | ||
| Scheme 4 Completion of the synthesis of diamine fragment 3. | ||
It is worth pointing out that the outcome of the Mitsunobu reaction (Route B, Scheme 4) is surprising considering the low acidity of both the primary alcohol 4 and dimethylamine starting materials, especially in the presence of the NHSOt-Bu moiety. Indeed, it was reported in the literature that tert-butylsulfinamines 13 can react intramolecularly with a proximate alcohol to form a five-membered pyrrolidine ring 14 (Scheme 5a).23 In our case, the formation of a four-membered azetidine ring should be more energetically demanding.24 Moreover, we could demonstrate (see supporting information†) that the N-methylated precursor 15 did not react under the Mitsunobu conditions with dimethylamine (Scheme 5b). Therefore, we assume that the NH bond favours the formation of the phosphonium intermediate, thereby allowing the subsequent nucleophilic substitution to take place, even with dimethylamine having a high pKa value. On the other hand, the formation of a phosphorane intermediate could not be ruled out.25
 | ||
| Scheme 5 Inter- versus intra-molecular amination under Mitsunobu conditions. | ||
Next, we embarked on a larger scale synthesis of diamine target 3 by optimizing our validated sequence, with special attention paid to minimize the number of purification procedures initially required at each reaction step (Scheme 6). Subsequent to the easy formation of sulphide 16 on a 2 kilogram scale, the acetal deprotection into aldehyde 8 was performed with H2SO4, in order to prevent the use of corrosive HCl. Keeping the green solvent MeTHF as the reaction media, the formation of imine 6 was conveniently carried out by means of a Dean–Stark distillation in the presence of the soft PPTS acid. This allowed the formation of sulfinimide 6 in a 68% crude yield (see supporting information†) on a kilogram scale through three telescoped steps (7 to 6). Though the purity of the product was estimated to be only 69% by means of HPLC analysis, this quality turned out to be sufficient for the subsequent steps. Disappointingly, a solvent screening revealed that the next Reformatsky reaction led to partial conversions in MeTHF solvent. Further optimisation and switching to THF demonstrated that imine 6 was completely transformed into amine 5 with a high diastereoisomeric ratio of 94:6, after a soft citric acid work-up to preserve the chiral auxiliary. It is worth noting that, according to a literature procedure,17d the activation of zinc metal by DIBAL-H is preferred to avoid the initially uncontrollable exothermicity during the Reformatsky's reagent formation. A column chromatography on silica gel improved the purity of product 5 from 68% to 82%, as estimated by HPLC analysis, which was found to be sufficient for the next step. The reduction of the ester group by Red-Al (5 to 4) and the subsequent Mitsunobu reaction were next successfully telescoped in the same solvent to furnish the crude amine 11. A silica gel column chromatography was required to remove the large amount of triphenylphosphine side product, and to allow the isolation of amine 11 in a good 64% yield and more than 99% purity, as measured by HPLC. Moreover, the isolation of the pure major diastereoisomer of 11 was secured at this stage. The chiral auxiliary was removed by HCl in methanol, and the corresponding diamine 3 was obtained in toluene solution after neutralization. Then, the final product 17 was conveniently isolated as a solid fumarate salt which furnished a pure material in 99.5% ee. It is worth noting that all attempts to perform the one-step deprotection of sulfinamine 11 by fumaric acid were unsuccessful.
 | ||
| Scheme 6 Scale-up synthesis of diamine building block 3 fumarate salt. | ||
Conclusions
A novel asymmetric synthesis of an enantiopure 1,3-diamine 3, a key fragment of potent Bcl-2/Bcl-xL protein inhibitor, was accomplished in seven linear steps. The two key steps involve both a highly diastereoselective aza-Reformatsky reaction on a chiral sulfinimine and a chemoselective Mitsunobu reaction allowing the introduction of the dimethylamine moiety. This laboratory synthesis of diamine 3 was demonstrated to be amenable to a larger-scale process up to the kilogram scale for some steps and required only two purifications by column chromatography. Both enantiomers of diamine 3 were shown to be available as useful building blocks of bioactive material.Acknowledgements
This work has been partially supported by INSA Rouen, Rouen University, CNRS, EFRD, Labex SynOrg (ANR-11-LABX-0029) and Région Haute-Normandie (CRUNCH network). We warmly thank J.-P. Lecouvé, the pilot plant team and the analytical team for their valuable support.Notes and references
- (a) N. N. Danial and S. J. Korsmeyer, Cell, 2004, 116, 205 CrossRef CAS; (b) J. C. Reed, Am. J. Pathol., 2000, 157, 1415 CrossRef CAS; (c) D. L. Vaux and S. J. Korsmeyer, Cell, 1999, 96, 245 CrossRef CAS.
- (a) E. R. McDonald III and W. S. El-Deiry, Death Recept. Cancer Ther., 2005, 1 Search PubMed; (b) D. W. Nicholson, Nature, 2000, 407, 810 CrossRef CAS PubMed; (c) S. W. Lowe and A. W. Lin, Carcinogenesis, 2000, 21, 485 CrossRef CAS PubMed; (d) B. A. Ponder, Nature, 2001, 411, 336 CrossRef CAS PubMed.
- (a) J. M. Adams and S. Cory, Science, 1998, 281, 1322 CrossRef CAS; (b) A. Gross, J. M. McDonnell and S. J. Korsmeyer, Genes Dev., 1999, 13, 1899 CrossRef CAS.
- (a) V. Kitrkin, S. Joos and M. Zornig, Biochim. Biophys. Acta, 2004, 1644, 229 CrossRef PubMed; (b) S. Cory and J. M. Adams, Nat. Rev. Cancer, 2002, 2, 647 CrossRef CAS PubMed; (c) D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57 CrossRef CAS.
- (a) J. C. Reed, Adv. Pharmacol., 1997, 41, 501 CAS; (b) J. M. Adams and S. Cory, Oncogene, 2007, 26, 1324 CrossRef CAS PubMed.
- (a) J. M. Adams and S. Cory, Oncogene, 2007, 26, 1324 CrossRef CAS PubMed; (b) P. Juin, O. Geneste, E. Raimbaud and J. A. Hickman, Biochim. Biophys. Acta, 2004, 1644, 251 CrossRef CAS PubMed.
- A. M. Petros, E. T. Olejniczak and S. W. Fesik, Biochim. Biophys. Acta, 2004, 1644, 83 CrossRef CAS PubMed.
- M. Sattler, H. Liang, D. Nettesheim, R. P. Meadows, J. E. Harlan, M. Eberstadt, H. S. Yoon, S. B. Shuker, B. S. Chang, A. J. Minn, C. B. Thompson and S. W. Fesik, Science, 1997, 275, 983 CrossRef CAS.
- (a) Y. Feng, X. Ding, T. Chen, L. Chen, F. Liu, X. Jia, X. Luo, X. Chen, K. Chen, H. Jiang, H. Wang, H. Liu and D. Liu, J. Med. Chem., 2010, 53, 3465 CrossRef CAS PubMed; (b) J. Wei, S. Kitada, M. F. Rega, J. L. Stebbins, D. Zhai, J. Cellitti, H. Yuan, A. Emdadi, R. Dahl, Z. Zhang, L. Yang, J. C. Reed and M. Pellecchia, J. Med. Chem., 2009, 52, 4511 CrossRef CAS PubMed; (c) G. Lessene, P. E. Czabotar and P. M. Colman, Nat. Rev. Drug Discovery, 2008, 7, 989 CrossRef CAS PubMed; (d) G. Tang, Z. Nikolovska-Coleska, S. Qiu, C.-Y. Yang, J. Guo and S. Wang, J. Med. Chem., 2008, 51, 717 CrossRef CAS PubMed; (e) G. Tang, C.-Y. Yang, Z. Nikolovska-Coleska, J. Guo, S. Quiu, R. Wang, W. Gao, G. Wang, J. Stuckey, K. Krajewski, S. Jiang, P. P. Roller and S. Wang, J. Med. Chem., 2007, 50, 1723 CrossRef CAS PubMed; (f) A. M. Petros, J. Dinges, D. J. Augeri, S. A. Baumeister, D. A. Betebenner, M. G. Bures, S. W. Elmore, P. J. Hajduk, M. K. Joseph, S. K. Landis, D. G. Nettesheim, S. H. Rosenberg, W. Shen, S. Thomas, X. Wang, I. Zanze, H. Zhang and S. W. Fesik, J. Med. Chem., 2006, 49, 656 CrossRef CAS PubMed.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. Ng, P. M. Nimmer, J. M. O'Connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. Wang, M. D. Wendt, H. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677 CrossRef CAS PubMed.
- (a) S. Barelier, J. Pons, O. Marcillat, J.-M. Lancelin and I. Krimm, J. Med. Chem., 2010, 53, 2577 CrossRef CAS PubMed; (b) P. J. Hajduk and J. A. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211 CrossRef CAS PubMed.
- C.-M. Park, M. Bruncko, J. Adickes, J. Bauch, H. Ding, A. Kunzer, K.-C. Marsh, P. Nimmer, A. R. Shoemaker, X. Song, S. K. Tahir, C. Tse, X. Wang, M. D. Wendt, X. Yang, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2008, 51, 6902 CrossRef CAS PubMed.
- (a) Y. Tanaka, K. Aikawa, G. Nishida, M. Homma, S. Sogabe, S. Igaki, Y. Hayano, T. Sameshima, I. Miyahisa, T. Kawamoto, M. Tawada, Y. Imai, M. Inazuka, N. Cho, Y. Imaeda and T. Ishikawa, J. Med. Chem., 2013, 56, 9635 CrossRef CAS PubMed; (b) H. Zhou, A. Aguilar, J. Chen, L. Bai, L. Liu, J. L. Meagher, C.-Y. Yang, D. McEachern, X. Cong, J. A. Stuckey and S. Wang, J. Med. Chem., 2012, 55, 6149 CrossRef CAS PubMed; (c) G. Lessene, P. E. Czabotar and P. M. Colman, Nat. Rev. Drug Discovery, 2008, 7, 989 CrossRef CAS PubMed.
- (a) M. Bruncko, T. K. Oost, B. A. Belli, H. Ding, M. K. Joseph, A. Kunzer, D. Martineau, W. J. McClellan, M. Mitten, S.-C. Ng, P. M. Nimmer, T. Oltersdorf, C.-M. Park, A. M. Petros, A. R. Shoemaker, X. Song, X. Wang, M. D. Wendt, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2007, 50, 641 CrossRef CAS PubMed; (b) M. D. Wendt, W. Shen, A. Kunzer, W. J. McClellan, M. Bruncko, T. K. Oost, H. Ding, M. K. Joseph, H. Zhang, P. M. Nimmer, S.-C. Ng, A. R. Shoemaker, A. M. Petros, A. Oleksijew, K. Marsh, J. Bauch, T. Oltersdorf, B. A. Belli, D. Martineau, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2006, 49, 1165 CrossRef CAS PubMed.
- (a) R. B. Biebold, T. Gero, P. Grover, S. Huang, S. Ioannidis, C. A. Ogoe, J. C. Saeh and J. G. Varnes, PCT Int. Appl., WO 2012017251, 2012; (b) O. J. Shah, Y. Shen, X. Lin, M. Anderson, X. Huang, J. Li and L. Li, PCT Int. Appl., WO 2011068863, 2011; (c) A. R. Kunzer, S. W. Elmore, L. Hexamer, C.-M. Park, A. J. Souers, G. M. Sullivan, G. T. Wang, X. Wang and M. D. Wendt, PCT Int. Appl., WO 2010083442, 2010; (d) S. W. Elmore, M. Bruncko and C.-M. Park, U.S. Pat. Appl. Publ., US 20050272744, 2005.
- For a recent insightful review, see: M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- (a) F. Grellepois, J. Org. Chem., 2013, 78, 1127 CrossRef CAS PubMed; (b) A. Sorochinsky, N. Voloshin, A. Markovsky, M. Belik, N. Yasuda, H. Uekusa, T. Ono, D. O. Berbasov and V. A. Soloshonok, J. Org. Chem., 2003, 68, 7448 CrossRef CAS PubMed; (c) D. D. Staas, K. L. Savage, C. F. Homnick, N. N. Tsou and R. G. Ball, J. Org. Chem., 2002, 67, 8276 CrossRef CAS PubMed; (d) M. J. Girgis, J. K. Liang, Z. Du, J. Slade and K. Prasad, Org. Process Res. Dev., 2009, 13, 1094 CrossRef CAS.
- (a) H. Peng, Y. Cheng, N. Ni, M. Li, G. Choudhary, H. T. Chou, C.-D. Lu, P. C. Tai and B. Wang, ChemMedChem, 2009, 4, 1457 CrossRef CAS PubMed; (b) H. Ishibashi, C. Kameoky, H. Iriyama, K. Kodama, T. Sato and M. Ikeda, J. Org. Chem., 1995, 60, 1276 CrossRef CAS.
- T. Scherkenbeck and K. Siegel, Org. Process Res. Dev., 2005, 9, 216 CrossRef CAS.
- J. Dekker, P. H. M. Budzelaar, J. Boersma and G. J. M. van des Kerk, Organometallics, 1984, 3, 1403 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974 CrossRef CAS.
- The more classical two-step approach, namely the N-alkylation of dimethylamine by means of mesylated or tosylated alcohol 4 was also abandoned because of eratic outcomes obtained during the scale-up process mainly due to the poor stability of these labile sulfonate ester intermediates.
- (a) I. Bosque, E. Bagdatli, F. Foubelo and J. C. Gonzalez-Gomez, J. Org. Chem., 2014, 79, 1796 CrossRef CAS PubMed; (b) K. N. Hahn, O. O. Fadeyi, H. P. Cho and C. W. Lindsley, Tetrahedron Lett., 2012, 53, 3577 CrossRef CAS PubMed.
- The Mitsunobu reaction conducted from the starting material 4 alone led after 1 hour to a complex mixture which failed to provide the desired product 11 upon subsequent addition of dimethylamine. This precludes the involvement of a four-membered ring azetine as a reactive intermediate. For the synthesis of stable N-tert-butylsulfinyl azetidines, see: C. Guérot, B. H. Tchitchanov, H. Knust and E. M. Carreira, Org. Lett., 2011, 13, 780 CrossRef PubMed.
- I. Mathieu-Pelta and S. A. Evans Jr, J. Org. Chem., 1992, 57, 3409 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: For procedures and compound characterisation. See DOI: 10.1039/c4ra07821g |
| ‡ These two researchers equally contributed to this project. |
| This journal is © The Royal Society of Chemistry 2014 |