
Solvothermal syntheses of lanthanide thiogermanates displaying three new structural moieties†
Rong-Qing Zhao,
Jian Zhou*,
Xing Liu*,
Li Zhang,
Qiuling Tang and
Xiao-Feng Tan
Key Laboratory of Green Synthesis and Applications, College of Chemistry, Chongqing Normal University, Chongqing, 401331, PR China. E-mail: Jianzhou888888@163.com; lx7507@163.com; Fax: +86-023-65419972; Tel: +86-023-65419972
First published on 19th August 2014
Abstract
A series of lanthanide thiogermanates [Ln(dien)3]2[Ge2S6]Cl2 [Ln = Pr (Ia), Sm (Ib), Gd (Ic), Dy (Id); dien = diethylenetriamine], [Er2(dien)4(μ-OH)2][Ge2S6] (II) and [Ho(trien)(en)GeS3(SH)] (III, trien = triethylenetetramine, en = ethylenediamine) have been hydrothermally synthesized and structurally characterized. The structures of Ia–d consist of isolated [Ln(dien)3]3+ cations, [Ge2S6]4− anions built up from the connection of two [GeS4] tetrahedra sharing a common edge and Cl− ions. II contains binuclear [Er2(dien)4(μ-OH)2]4+ cations constructed by the linkage of [Er(dien)2]3+ ions and –OH bridging groups, and [Ge2S6]4− anions. III contains neutral holmium-centred complexes, where the unusual protonated tetrahedral anion [GeS3(SH)]3− acts as a chelating ligand to complex the [Ho(en)(trien)]3+ cation. A systematic investigation of six lanthanide thiogermanates and four reported compounds revealed that both the well-known lanthanide contraction and different chelating organic amines have a significant influence on the formation of lanthanide thiogermanates under solvothermal conditions. Density functional theory calculation for III has also been performed and the absorption edges of all compounds have been investigated by UV-vis spectroscopy.
Introduction
Solvothermal synthesis of new chalcogenidogermanates has received increasing attention since 1989,1 due to their fascinating architectures and potential applications in gas separation, ion conductivity, and photocatalysis.2 Among these compounds, the thiogermanates have been well studied, fundamental [GeS4] tetrahedral units or combination of [GeS4] and [MS4] (M = Ga, In) tetrahedral units can condense into the [MxGe4−xS10] (x = 0–3) supertetrahedral units (T2) as secondary building units, which further self-assemble into a variety of open framework thiogermanates.2d The T2 unit and other [MSx] polyhedra (M = transition-metal ions) are interconnected to form the various 3-D network structures with pores or cavities.3 Protonated nonchelating amines or tetraalkyl-ammonium ions as structure-directing agents are commonly retained within pore or cavity spaces. The [GeS4] unit can also combine with acentric [SbSn] (n = 3, 4) polyhedra via corner sharing of vertex S atoms to produce 1-D, 2-D, and 3-D thiogermanate-thioantimonates.4 When transition metal complexes substitute for nonchelating amines or tetraalkyl-ammonium ions, thiogermanate anions are strongly dominated by [Ge2S6]4− or [Ge4S10]4− moieties.5 Notably, the noncondensed [GeS4]4− mode in organic hybrid thiogermanates is very scarce, and appear to be limited to that of [VO(dien)]2GeS4,6 [Ln(dien)2(GeS3(SH))]n (Ln = La, Nd),7 and [Na(H2O)3]-[Cr(en)3]2[GeS3(OH)]2[Cr(en)2(GeS4)],8 where the [GeS4]4− ion is stabilized by coordinating to metal complex. More interestingly, the cations of these thiogermanates are either protonated amine or transition metal complex cations,3,5,6,8 but the thiogermanates with lanthanide-containing counter cation prepared under mild solvothermal conditions have been still a less explored area.The integration of lanthanide (Ln) metals in chalcogenide frameworks can lead to new materials with interesting semiconducting, magnetic, luminescent and infrared nonlinear optical properties.9 These materials have traditionally been prepared by flux methods at high temperature, as exemplified by La2Ga2GeS8,10 Ba3AGa5Se10Cl2 (A = Cs, Rb, K),11 and Ba23Ga8Sb2S38.12 However, the synthesis of lanthanide chalcogenides modified with organic components are hampered by the heat, water, oxygen and light sensitivity of these materials, and the difficulty of soft Lewis basic Q2− ligands (Q = S, Se, Te) coordinating to the hard Lewis acidic Ln3+ ions via Ln–Q bonds in the solvents.13 Recently, we have attempted the preparation of lanthanide thiogermanates in chelating amine solutions by the solvothermal methods allowing access to thermodynamically metastable phases, and prepared four organic hybrid lanthanide thiogermanates, [Eu(dien)3]2[Ge2S6]Cl,14 [Y2(tepa)2(μ-OH)2(μ-Ge2S6)](tepa)0.5·H2O (tepa = tetraethylenepentamine),14 and [Ln(dien)2(GeS3(SH))]n (Ln = La, Nd),7 where the soft Lewis basic [Ge2S6]4− and [GeS3(SH)]3− units act as ligands to the hard Lewis acidic Ln3+ ions. To investigate systematically influence of the well-known lanthanide contraction and different chelating organic amines on the formation of lanthanide thiogermanates, we started to explore the system LnCl3/GeO2/S/amine under solvothermal conditions, and successfully synthesized a series of organic hybrid lanthanide thiogermanates, [Ln(dien)3]2[Ge2S6]Cl2 [Ln = Pr (Ia), Sm (Ib), Gd (Ic), Dy (Id)], [Er2(dien)4(μ-OH)2][Ge2S6] (II) and [Ho(trien)(en)GeS3(SH)] (III). I and II contain isolated [Ge2S6]4− anions with lanthanide complexes as counterions, while III consists of neutral holmium-centred complex with noncondensed tetrahedral anion [GeS3(SH)]3− as a chelating ligand to complex [Ho(en)(trien)]3+ cation.
Experimental section
General remarks
All analytical grade chemicals were obtained commercially and used without further purification. Elemental analyses (C, H, and N) were performed using a PE2400 II elemental analyzer. The UV/Vis spectra were recorded at room temperature using a computer-controlled PE Lambda 900 UV/Vis spectrometer equipped with an integrating sphere in the wavelength range of 200–1250 nm. FT-IR spectra were recorded with a Nicolet Magna-IR 550 spectrometer in dry KBr disks in the 4000–400 cm−1 range. Powder XRD patterns were collected on a D/MAX-3C diffractometer using graphite-mono-chromatized CuKa radiation (λ = 1.5406 Å).Crystal structure determination
Single-crystal X-ray diffraction data for all compounds were recorded on a Rigaku Mercury CCD diffractometer using a ω-scan method with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 296(2) K to a maximum 2θ value (52.00°). Absorption corrections were applied using multi-scan technique. The structures of all compounds were solved by Direct Methods (SHELXS-97)15 and refined by full-matrix least-squares techniques using the SHELXL-97 program.16 Non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms bonded to C and N atoms were positioned with idealized geometry and a riding model was used. H atom associated with –SH group in III was located from the difference Fourier map. Relevant crystal and collection data parameters and refinement results can be found in Table 1.| Ia | Ib | Ic | Id | II | III | |
|---|---|---|---|---|---|---|
| Formula | C12H39ClGeN9PrS3 | C12H39ClGeN9S3Sm | C12H39ClGdGeN9S3 | C12H39ClDyGeN9S3 | C16H54Er2Ge2N12O2S6 | C8H27GeHoN6S4 |
| Fw | 654.70 | 664.15 | 671.04 | 676.29 | 1118.87 | 573.18 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n | P21/n | P21/n | P21/n | P21/c |
| a, Å | 11.637(3) | 11.532(3) | 11.5484(14) | 11.5035(11) | 11.710(2) | 9.7755(15) |
| b, Å | 14.143(3) | 14.423(4) | 14.6767(17) | 14.6453(14) | 11.318(2) | 13.4516(19) |
| c, Å | 15.120(3) | 14.573(4) | 14.4271(17) | 14.3396(13) | 13.548(2) | 14.388(2) |
| β, deg | 98.149(5) | 97.105(5) | 96.332(2) | 96.178(2) | 97.635(3) | 99.264(2) |
| V, Å3 | 2463.5(10) | 2405.3(10) | 2430.4(5) | 2401.8(4) | 1779.6(5) | 1867.3(5) |
| Z | 4 | 4 | 4 | 4 | 2 | 4 |
| T, K | 296(2) | 296(2) | 296(2) | 296(2) | 296(2) | 296(2) |
| Calcd density, Mg m−3 | 1.765 | 1.834 | 1.834 | 1.870 | 2.088 | 2.039 |
| Abs.coeff., mm−1 | 3.555 | 4.056 | 4.327 | 4.729 | 6.728 | 6.262 |
| F(000) | 1320 | 1332 | 1340 | 1348 | 1092 | 1120 |
| 2θ (max), deg | 50.20 | 50.20 | 46.78 | 50.20 | 50.20 | 50.18 |
| Total reflns collected | 17095 | 16607 | 20881 | 16686 | 12194 | 13209 |
| Unique reflns | 4249 | 4206 | 5982 | 4237 | 3110 | 3310 |
| No. of param. | 244 | 244 | 244 | 244 | 185 | 184 |
| R1[I > 2σ(I)] | 0.0235 | 0.0245 | 0.0213 | 0.0573 | 0.0171 | 0.0513 |
| wR2 (all data) | 0.0570 | 0.0667 | 0.0640 | 0.1568 | 0.0418 | 0.1331 |
Results and discussion
Synthetic aspects
Solvothermal techniques have proven to be an extremely effective method for the preparation of new chalcogenometalates, which overcome the difficulties associated with differential solubilities of organic and inorganic starting materials.17 The crystals of Ia–d, [Eu(dien)3]2[Ge2S6]Cl2,14 and II were obtained in the presence of dien. Dien preferably coordinate to Ln3+ ions to form isolated complex cations [Ln(dien)3]3+ or [Ln2(dien)4(μ-OH)2]4+, which prevent the lanthanide complexes from bonding with S atom of thiogermanate anion. Penta-dentate chelating amine coordinate to Ln3+ ion to form unsaturated lanthanide complex that can effectively incorporate into chalcogenido-germanate frameworks to product novel organic hybrid lanthanide chalcogenido-germanates with diverse structures.14,18 Mixed chelating ammines can also lead to the formation of unsaturated lanthanide complex, as exemplified by [Ho(trien)(en)GeS3(SH)] (III). These results demonstrated that different chelating organic amines as structure-directing agents have a significant influence on the formation of lanthanide chalcogenidogermanate under solvothermal conditions (Scheme 1). | ||
| Scheme 1 View of the structural moieties of lanthanide thiogermanates with different chelating organic amine under solvothermal conditions. | ||
Crystal structure
Description of the structures of Ia–d. Ia, Ib, Ic and Id are isomorphic, so we only discussed the structure of Ia. Ia crystallizes in monoclinic centrosymmetric space group of P21/n and contains one half [Ge2S6]4− anion, one complete [Pr(dien)3]3+ and one Cl− ion in the asymmetric unit (Fig. 1a). The [Pr(dien)3]3+ cation needs additional one Cl− ion to compensate its positive charge. The dimeric [Ge2S6]4− anion exhibits crystallographic Ci symmetry, which is formed by two edge-sharing [GeS4] tetrahedra with four terminal S atoms. The Ge–Sb (b = bridging) bond distances (2.2596(10)-2.2803(10) Å) are slightly longer than the Ge–St (t = terminal) bond distances (2.1518(10)–2.1627(10) Å). The distorted tetrahedral [GeS4] geometry can be seen from the S–Ge–S bond angles (92.09(4)–113.78(5)°), which deviate significantly from the ideal value of 109.5°. The Pr3+ ion is coordinated by 9 N atoms from three dien ligands. The [EuN9] polyhedron can be described as a distorted tri-capped trigonal prism (Fig. 1b). The Ln–N bond lengths are 2.651(3)–2.733(3) Å for Pr–N, 2.599(4)–2.668(4) Å for Sm–N, 2.577(3)–2.661(3) Å for Gd–N and 2.544(6)–2.629(6) Å for Dy–N, and are consistent with those in other amino Ln3+ complexes.19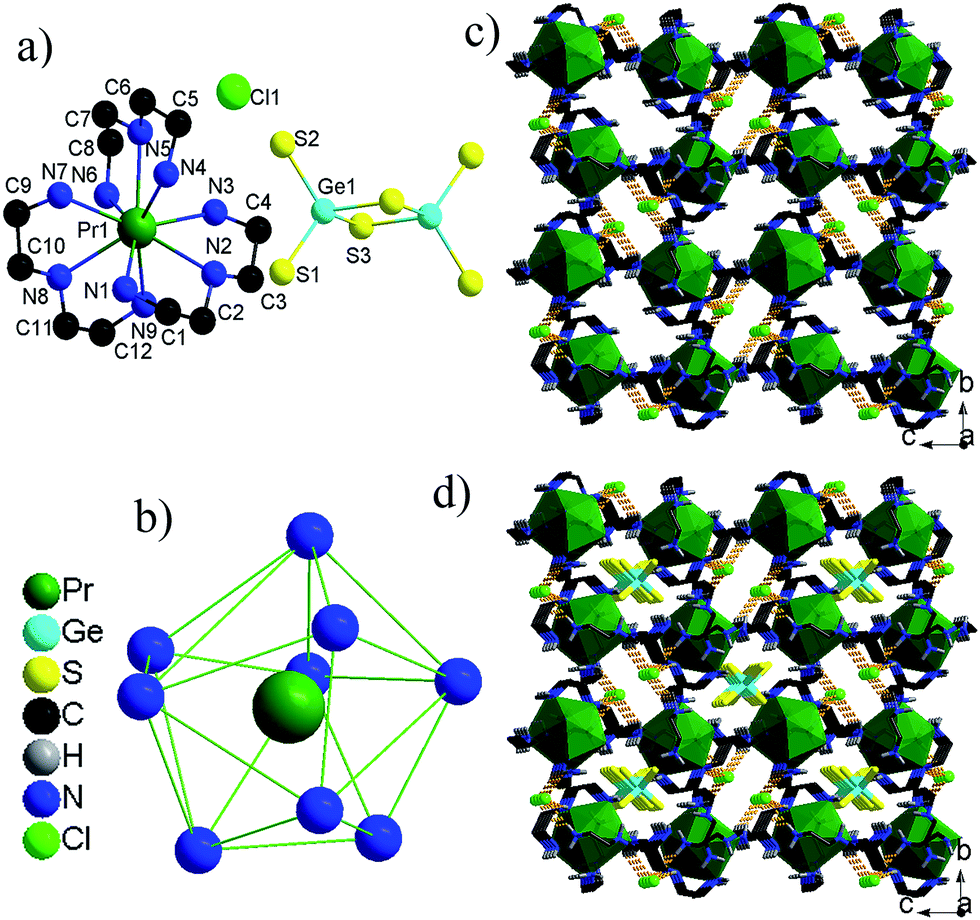 | ||
| Fig. 1 (a) Molecular structure of Ia [all H atoms are omitted for clarity]. (b) The coordination environments of Pr3+ ion. (c) The stack of [Pr(dien)3]3+ and Cl− ions in Ia, showing 1-D tunnel-like features. (d) The 3-D supermolecular network in Ia. H atoms bonded to C atoms are omitted for clarity. | ||
The [Pr(dien)3]3+ cations adopt the NaCl-type stacking mode, (Fig. 1c). The shortest Pr⋯Pr distance is 9.44 Å. Cl− ions are involved in intermolecular N–H⋯Cl hydrogen bonding with the –NH2 groups of adjacent [Pr(dien)3]3+ ions resulting in a 2-D layer (Fig. S1†). These layers are arranged in parallel with an interlayer distance of about 11.64 Å, leading to the formation of 1-D tunnel-like features that are filled by the [Ge2S6]4− anions (Fig. 1d). There are a lot of short intermolecular N–H⋯S interactions that play an important role in stabilizing 1 in the solid state.
Description of the structure of II. II crystallizes in monoclinic centrosymmetric space group of P21/n with two formula units in the unit cell. II consists of a discrete dimeric [Ge2S6]4− anion and a charge compensating binuclear [Er2(dien)4(μ-OH)2]4+ complex cation. Two [Er(dien)2]3+ groups are joined by two [μ2-OH] bridging groups to form a centrosymmetric dinuclear complex cation with a [Er2O2] rhomboidal core. The Er⋯Er separation is 3.7231(5) Å, which is similar to that observed in other polynuclear erbium complexes.20 The Er3+ center is in a 8-fold coordination of six N atoms of two dien ligands and two O atoms of two [μ2-OH] groups. The [ErN6O2] polyhedron can be described as a distorted bicapped trigonal prism. The Er–O (2.216(2)–2.251(2) Å) and Er–N (2.466(3)–2.563(3) Å) bond lengths are compared with those reported for [Er2(en)6(μ2-OH)2][Sn2S6].20 The dien ligands in [Er2(dien)4(μ-OH)2]4+ ion display two different conformation, namely facial and U-shape, while the dien ligands of [Ln(dien)3]3+ ions in Ia–d show only one type of facial conformation. The interatomic distances and angles in the [Ge2S6]4− anion are similar to those in Ia–d and other discrete [Ge2S6]4− anions.5 There are many important N–H⋯S and O–H⋯S hydrogen bonds between the [Ge2S6]4− anions and [Er2(dien)4(μ-OH)2]4+ cations. By these hydrogen-bond interactions, the [Ge2S6]4− anions and Er2(dien)4(μ-OH)2]4+ cations are assembled into a 3-D hydrogen-bond network structure (Fig. 2b).
 | ||
| Fig. 2 (a) Molecular structure of II [H atoms bonded to C/N atoms are omitted for clarity]. (b) 3-D hydrogen bonding network structure [H atoms bonded to C atoms are omitted for clarity]. | ||
Description of the structure of III. III crystallizes in monoclinic centrosymmetric space group of P21/c with four formula units in the unit cell. The Ho3+ ion is coordinated by one teta ligand and one bidentate [η2-GeS3(SH)] chelating anion to give a neutral molecule [Ho(trien)(en)GeS3(SH)] with an Ho⋯Ge distance of 3.521(1) Å (Fig. 3a). The polyhedron [HoN6S2] can be described as a distorted bicapped trigonal prism (Fig. 3b). The Ho–N (2.474(7)–2.541(6) Å) and Ho–S (2.7927(19)–2.833(2) Å) bonds are comparable to those in [Ho(1,10-phen)L3] (L = N,N-diethylithiocarbato).21 The [GeS3(SH)]3− anion show a distorted tetrahedral geometry. The Ge1–S4 bond (2.317(3) Å) is significantly longer than other Ge–S bonds (2.163(2)–2.201(2) Å), showing that S4 is protonated. A very weak band at 2575 cm−1, assigned to the S–H stretching vibration, confirms the presence of an –SH group. The –SH group has also been reported in the other sulfides [Ni(tepa)]2[In4S7(SH)2]·H2O22 and [Ln(dien)2(μ-η1,η2-GeS3(SH))]n (Ln = La, Nd).7
 | ||
| Fig. 3 (a) Molecular structure of III [all H atoms are omitted for clarity]. (b) The coordination environments of Ho3+ ion. (c) Part of the crystal structure of III, showing the formation of a (100) sheet constructed from N–H⋯S hydrogen bonds. (d) 3-D hydrogen bond network structure. H atoms bonded to C atoms are omitted for clarity. | ||
There are a lot of weak N–H⋯S hydrogen bonds between –NH groups and S atoms of [GeS3(SH)]3− anions. The [Ho(trien)(en)-GeS4H] molecules are connected via N–H⋯S hydrogen bonds to form lays parallel to the (100) plane (Fig. 3c) with an interlayer distance of about 9.78 Å. Further interactions via N–H⋯S H-bonds gives a 3-D hydrogen bonding network (Fig. 3d).
The [GeS4] tetrahedra under solvothermal conditions exhibit a characteristic tendency to condense via corner- or edge-bridging forming dimeric [Ge2S6]4− or [Ge4S10]4− moieties with transition metal complexes as counterions.5 But simply noncondensed [GeS4]4− or [GeS3(SH)]3− anion combined with lanthanide complexes is very scarce, and appear to be limited to that of [Ln(dien)2(GeS3(SH))]n (Ln = La, Nd),7 where the [GeS3(SH)]3− anion acts as a [μ-η1,η2-GeS3(SH)] bridging ligand to [Ln(dien)2]3+ group to form a 1-D coordination polymer. The [GeS3(SH)]3− anion in III acting as bidentate chelating ligand is the second example of noncondensed [GeS4]4− or [GeS3(SH)]3− species with lanthanide complexes under solvothermal conditions. Moreover, these organic hybrid lanthanide thiogermanates made by using solvothermal techniques usually contain only one type of organic amine, but different organic amines as chelating ligands and structure-directing agents have not been documented to date. Hence, III is the only known lanthanide thiogermanate combined with two different organic ligands.
Lanthanide contraction effect on the structures of lanthanide thiogermanates
Lanthanide contraction is a term used in chemistry to describe the steady decrease in the size of the atoms and ions of the lanthanide elements with increasing atomic number, as shown in compounds with coordination numbers nine for the earlier lanthanide(III) ions and eight for the later ones in aqueous solution.23 The dien ligand prefers to coordinate to lighter lanthanide ions to form [Ln(dien)3]3+ ions [Ln = Pr (Ia), Sm (Ib), Gd (Ic), Dy (Id) and Eu14], and to coordinate to heavier ones to give saturated binuclear [Ln2(dien)4(μ-OH)2]4+ (Ln = Er(II)). The saturated [Ln(dien)3]3+ or [Ln2(dien)4(μ-OH)2]4+ ions only act as [Ge2S6]4− anionic counterion. Evidently, two types of lanthanide complex cations are related to the Ln3+ ion size. To complete a coordination number of nine for both La3+ and Nd3+ ions, and eight for Ho3+ ion, the [GeS3(SH)]3− anion coordinates to unsaturated [Ln(dien)2]3+ or [Ho(trien)(en)]3+ ions forming 1-D neutral chains [Ln(dien)2(GeS3(SH))]n (Ln = La, Nd)7 and molecular [Ho(trien)(en)GeS3(SH)] (III). These results demonstrated that lanthanide contraction has a significant influence on the formation of lanthanide thiogermanates under solvothermal conditions.Optical properties
The solid-state UV/Vis absorption spectra of all compounds measured at room temperature are shown in Fig. 4 and S2.† The absorption edges are 2.35 eV for Ia, 2.36 eV for Ib, 2.36 eV for Ic, 3.59 eV for Id, 3.23 eV for II, and 3.54 eV for III, which are compared with values of other thiogermanates (2.10–3.53 eV), such as [Mn(en)3]GeSb2S6 (2.10 eV, en = ethylenediamine),4c [Co(dien)2]2GeSb4S10 (2.36 eV),4c [Eu(dien)3]2[Ge2S6]Cl2 (2.27 eV),14 [Y2(tepa)2(μ-OH)2(μ-Ge2S6)](tepa)0.5·H2O (2.82 eV, tepa = tetraethylenepentamine),14 [M(dap)3]4Ge4S10Cl4 [M = Co (3.21 eV) and Ni (3.31 eV), dap = diaminopropane],5b [Ge3S6Zn(H2O)S3Zn(H2O)][(Zn(tren)(H2O)] (3.40 eV, tren = tris(2-aminoethyl)amine)24 and (trenH2)2[Ge2S6] (3.53 eV).14 Moreover, there are several absorptions characteristic of the f–f transitions of Pr3+ {1.22 eV (3H4 → 1G4) and 2.07 eV (3H4 → 1D2)}, Sm3+ {1.00 eV (6H5/2 → 6F7/2), 1.14 eV (6H5/2 → 6F9/2), 1.29 eV (6H5/2 → 6F11/2)}, Dy3+ {1.13 eV (6H15/2 → 6H7/2 + 6F9/2), 1.37 Ev (6H15/2 → 6F7/2), 1.53 eV (6H15/2 → 6F5/2)}, Ho3+ {1.27 eV (5I8 → 5I6), 1.89 eV (5I8 → 5I5), 2.38 eV (5I8 → 5S2 + 5F4), 2.55 eV (5I8 → 5F3), 2.74 eV (5I8 → 5G6), 3.05 eV (5I8 → 5G5 + 3G5)} and Er3+ {1.07 eV (4I15/2 → 4F11/2), 1.93 eV (4I15/2 → 4F9/2), 2.30 eV (4I15/2 → 4S3/2), 2.54 eV (4I15/2 → 4F7/2), 2.73 eV (4I15/2 → 4F5/2), 2.98 eV (4I15/2 → 4F3/2), 3.43 eV (4I15/2 → 2H9/2)} observed, which are agreement with the complicated UV-vis spectra of previously reported molecular lanthanide complexes.25 No such band could be observed for Id. | ||
| Fig. 4 The solid-state UV-vis absorption spectra of Ia, II and III. | ||
Theoretical studies
The band structure and density of states (DOS) of III were theoretically calculated using the computer code CASTEP. The valence bands (VBs) are dominated by the S 3p block, while the conduction bands (CBs) are mainly derived from Ho 4f states with a small contribution from Ge 4s orbitals (Fig. 5). Notably, the contributions from N and C atoms near the Fermi level are almost neglectable. The electronic absorption responsible for the optical gap is likely an electronic transfer excitation of S 3p to Ho 4f orbital electrons. The state energies of the lowest CBs and the highest VBs are located in the same Gamma point (Fig. S3†), and computational band gap of III is about 1.55 eV. Such value is smaller than the measured optical band gap, which may be related to the underestimation of the band gap by the DFT method.26 | ||
| Fig. 5 The total density-of-states and partial density-of-states for III. The Fermi level is set at 0 eV (dotted line). | ||
Conclusions
Three structural types of lanthanide thiogermanates were obtained from LnCl3/GeO2/S/amine system by mild solvothermal reaction. Although some thiogermanates combinated with metal complexes have been reported, their anions are strongly dominated by [Ge2S6]4− or [Ge4S10]4− moieties. III contains simple tetrahedral [GeS3(SH)]3− acting as a chelating ligand to complex [Ho(en)(trien)]3+ cation, which is the rare example of noncondensed [GeS4]4− or [GeS3(SH)]3− species under solvothermal conditions. The above observations on the organic hybrid lanthanide thiogermanates could be helpful for a full complete comprehension of the coordination chemistry of Ln3+ ions.Acknowledgements
This work was supported by the NNSF of china (no. 21163022), the NSF of Chongqing municipality (no. cstc2014jcyjA50002), the Key Project of Chinese Ministry of Education (no. 212132), and Program for Excellent Talents in Chongqing Higher Education Institutions. The authors are also grateful to Chongqing Normal University for financial support (13XLZ07).Notes and references
- R. L. Bedard, S. T. Wilson, L. D. Vail, J. M. Bennett and E. M. Flanigen, in Zeolites: Facts, Figures, Future. Proceedings of the 8th International Zeolite Conference, ed. P. A. Jacobs and R. A. van Santen, Elsevier, Amsterdam, 1989, pp. 375–387 Search PubMed.
- (a) Z. Zhang, J. Zhang, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2008, 130, 15238–15239 CrossRef CAS PubMed; (b) S. Bag and M. G. Kanatzidis, J. Am. Chem. Soc., 2008, 130, 8366–8376 CrossRef CAS PubMed; (c) K. K. Rangan, P. N. Trikalitis and M. G. Kanatzidis, J. Am. Chem. Soc., 2000, 122, 10230–10231 CrossRef CAS; (d) N. Zheng, X. Bu, B. Wang and P. Feng, Science, 2002, 298, 2366–2369 CrossRef CAS PubMed.
- (a) O. Achak, J. Y. Pivan, M. Maunaye, M. Loüer and D. Loüer, J. Solid State Chem., 1996, 121, 473–478 CrossRef CAS; (b) K. Tan, Y. Ko, J. B. Parise and A. Darovsky, Chem. Mater., 1996, 8, 448–453 CrossRef CAS; (c) C. L. Bowes, W. U. Huynh, S. J. Kirkby, A. Malek, G. A. Ozin, S. Petrov, M. Twardowski and D. Young, Chem. Mater., 1996, 8, 2147–2152 CrossRef CAS; (d) C. L. Cahill and J. B. Parise, Chem. Mater., 1997, 9, 807–811 CrossRef CAS; (e) C. L. Cahill, Y. Ko, J. C. Hanson, K. Tan and J. B. Parise, Chem. Mater., 1998, 10, 1453–1458 CrossRef CAS; (f) K. Tan, A. Darovsky and J. B. Parise, J. Am. Chem. Soc., 1995, 117, 7039–7040 CrossRef; (g) O. M. Yaghi, Z. Sun, D. A. Richardson and T. L. Groy, J. Am. Chem. Soc., 1994, 116, 807–808 CrossRef CAS; (h) M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 397, 681–684 CrossRef CAS PubMed.
- (a) M. Feng, D. Kong, Z. Xie and X. Huang, Angew. Chem., Int. Ed., 2008, 47, 8623–8626 CrossRef CAS PubMed; (b) M. Feng, W. Xiong, D. Ye, J. Li and X. Huang, Chem.–Asian J., 2010, 5, 1817–1823 CrossRef CAS PubMed; (c) J. Zhou, L. An, X. Liu, L. Huang and X. Huang, Dalton Trans., 2011, 40, 11419–11424 RSC; (d) J. Zhou, X. Liu, G. Liang, W. Liang, F. Hu and L. Zhu, Inorg. Chem. Commun., 2013, 27, 92–96 CrossRef CAS PubMed; (e) M. Feng, C. Hu, K. Wang, C. Du and X. Huang, CrystEngComm, 2013, 15, 5007–5011 RSC; (f) A. V. Powell and R. Mackay, J. Solid State Chem., 2011, 184, 3144–3149 CrossRef CAS PubMed.
- (a) J. Liang, J. Zhao, W. Tang, Y. Zhang and D. Jia, Inorg. Chem. Commun., 2011, 14, 1023–1026 CrossRef CAS PubMed; (b) F. Zhang, X. Liu, J. Zhou, X. Yin and J. He, Monatsh. Chem., 2011, 142, 763–768 CrossRef CAS; (c) G. Liu, J. Lin, Z. Xu, Z. Liu, G. Guo and J. Huang, Cryst. Growth Des., 2011, 11, 3318–3322 CrossRef CAS; (d) D. Jia, J. Dai, Q. Zhu, L. Cao and H. Lin, J. Solid State Chem., 2005, 178, 874–881 CrossRef CAS PubMed; (e) G. Liu, G. Guo, M. Wang, L. Cai and J. Huang, J. Mol. Struct., 2010, 983, 104–111 CrossRef CAS PubMed; (f) F. Zhang, X. Yin, X. Liu and J. Zhou, Z. Anorg. Allg. Chem., 2011, 637, 1388–1393 CrossRef CAS PubMed; (g) X. Liu, J. Zhou, J. He and Z. Huang, Z. Naturforsch., 2011, 66b, 659–663 CrossRef PubMed.
- J. Wang, C. Näther, J. Djamil and W. Bensch, Z. Anorg. Allg. Chem., 2012, 638, 1452–1456 CrossRef CAS PubMed.
- J. Zhou, R. Li, X. Ling, R. Chen, F. Hu and Y. Zeng, Dalton Trans., 2013, 42, 1961–1964 RSC.
- M. Melullis, M. K. Brandmayer and S. Dehnen, Z. Anorg. Allg. Chem., 2006, 632, 64–72 CrossRef CAS PubMed.
- (a) M.-C. Chen, L.-H. Li, Y.-B. Chen and L. Chen, J. Am. Chem. Soc., 2011, 133, 4617–4624 CrossRef CAS PubMed; (b) M. F. Churbanov, I. V. Scripachev, V. S. Shiryaev, V. G. Plotnichenko, S. V. Smetanin, E. B. Kryukova, Y. N. Pyrkov and B. I. Galagan, J. Non-Cryst. Solids, 2003, 326–327, 301–305 CrossRef; (c) D. T. Tonchev, C. J. Haugen, R. G. DeCorby, J. N. McMullin and S. O. Kasap, J. Non-Cryst. Solids, 2003, 326/327, 364–368 CrossRef; (d) D. Wruck, A. Klimakow, B. Rau and F. Henneberger, Semicond. Sci. Technol., 2001, 16, 885–888 CrossRef CAS; (e) T. Schmidt, G. Mueller, L. Spanhel, K. Kerkel and A. Forchel, Chem. Mater., 1998, 10, 65–71 CrossRef CAS.
- M. Chen, P. Li, L. Zhou, L. Li and L. Chen, Inorg. Chem., 2011, 50, 12402–12404 CrossRef CAS PubMed.
- P. Yu, L. Zhou and L. Chen, J. Am. Chem. Soc., 2012, 134, 2227–2235 CrossRef CAS PubMed.
- M. Chen, L. Wu, H. Lin, L. Zhou and L. Chen, J. Am. Chem. Soc., 2012, 134, 6058–6060 CrossRef CAS PubMed.
- (a) A. Kornienko, S. Banerjee, G. A. Kumar, R. E. Riman, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2005, 127, 14008–14014 CrossRef CAS PubMed; (b) J. Zhou, X. Liu, L. An, F. Hu, W. Yan and Y. Zhang, Inorg. Chem., 2012, 51, 2283–2290 CrossRef CAS PubMed; (c) J. Zhou, F. Hu, L. An, X. Liu and C. Meng, Dalton Trans., 2012, 41, 11760–11764 RSC; (d) J. Zhou, L. An, F. Hu, X. Liu, R. Zhao and J. Lin, CrystEngComm, 2012, 14, 5544–5551 RSC; (e) J. Zhou, X. Liu, R. Li, L. An, F. Hu, R. Chen and Y. Wei, Dalton Trans., 2013, 42, 11155–11162 RSC.
- X. Liu, F. Hu, J. Zhou, L. An, D. Liang and J. Lin, CrystEngComm, 2012, 14, 3464–3468 RSC.
- (a) G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- (a) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) J. Zhou, J. Dai, G. Bian and C. Li, Coord. Chem. Rev., 2009, 253, 1221–1247 CrossRef CAS PubMed; (b) W. S. Sheldrick and M. Wachhold, Angew. Chem., Int. Ed. Engl., 1997, 36, 206–224 CrossRef CAS PubMed.
- R. Chen, W. Tang, J. Liang, W. Jiang, Y. Zhang and D. Jia, Dalton Trans., 2012, 41, 12439–12445 RSC.
- (a) D. Jia, Q. Zhao, Y. Zhang, J. Dai and J. Zuo, Inorg. Chem., 2005, 44, 8861–8867 CrossRef CAS PubMed; (b) D. Jia, Q. Zhu, J. Dai, W. Lu and W. Guo, Inorg. Chem., 2005, 44, 819–821 CrossRef CAS; (c) Q. Zhao, D. Jia, Y. Zhang, L. Song and J. Dai, Inorg. Chim. Acta, 2007, 360, 1895–1901 CrossRef CAS PubMed.
- D. Jia, A. Zhu, J. Deng and Y. Zhang, Z. Anorg. Allg. Chem., 2007, 633, 1246–1250 CrossRef CAS PubMed.
- M. D. Regulacio, N. Tomson and S. L. Stoll, Chem. Mater., 2005, 17, 3114–3121 CrossRef CAS.
- J. Zhou, G. Bian, Y. Zhang, Q. Zhu, C. Li and J. Dai, Inorg. Chem., 2007, 46, 6347–6352 CrossRef CAS PubMed.
- C. Cossy, A. C. Barnes, J. E. Enderby and A. E. Merbach, J. Chem. Phys., 1989, 90, 3254–3259 CrossRef CAS PubMed.
- N. Zheng, X. Bu and P. Feng, Chem. Commun., 2005, 2805–2807 RSC.
- (a) K. Teshima, S. Lee, N. Shikine, T. Wakabayashi, K. Yubuta and T. S. S. Oishi, Cryst. Growth Des., 2011, 11, 995–999 CrossRef CAS; (b) A. F. Silva, D. S. Moura, A. S. Gouveia-Neto, E. A. Silva Jr, L. A. Bueno, E. B. Costa and E. N. Azevedo, Opt. Commun., 2011, 284, 4501–4503 CrossRef PubMed; (c) G. Lakshminarayana and J. Qiu, Phys. B, 2009, 404, 1169–1180 CrossRef CAS PubMed; (d) B. Karmakar, J. Solid State Chem., 2005, 178, 2663–2672 CrossRef CAS PubMed.
- (a) C. M. I. Okoye, J. Phys.: Condens. Matter, 2003, 15, 5945–5958 CrossRef CAS; (b) H.-J. Zhao, L.-H. Li, L.-M. Wu and L. Chen, Inorg. Chem., 2010, 49, 5811–5817 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data in CIF format, additional figure, XRD data, and theoretical calculation. CCDC 1005128–1005132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra07812h |
| This journal is © The Royal Society of Chemistry 2014 |