
Influence of element doping on La–Mn–Cu–O based perovskite precursors for methanol synthesis from CO2/H2
Haijuan Zhanab,
Feng Lia,
Peng Gaoc,
Ning Zhao*a,
Fukui Xiaoa,
Wei Weiac and
Yuhan Sunad
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, South Taoyuan Road 27#, Taiyuan 030001, People's Republic of China. E-mail: zhaoning@sxicc.ac.cn; Fax: +86-351-041153; Tel: +86-351-4049612
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
cCenter for Greenhouse Gas and Environmental Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201203, People's Republic of China
dCAS Key Laboratory of Low-carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201203, People's Republic of China
First published on 25th September 2014
Abstract
Doped La–M–Mn–Cu–O based (M = Ce, Mg, Y, Zn) perovskite materials were prepared by the sol–gel method and characterized by XRD, N2-adsorption, ICP-OES, SEM, TPR, N2O-adsorption, XPS and TPD techniques. Upon the introduction of the fourth elements, all the samples keep the stable LaMnO3 perovskite structure, and part of the copper species is separated from the perovskite lattice. More structure defects, lower reduction temperature and better low-temperature H2 adsorption on the unit surface area are observed. In the application for methanol synthesis from CO2/H2, the Zn doped catalyst showed better performance which may because the strength of the weak basic sites play a significant role on methanol selectivity and the amount of H2 adsorbed on the unit surface area is the key for CO2 conversion.
1. Introduction
Perovskite-type oxides with the general formula of ABO3 has received much attention because of their important physical properties such as ferro-, piezo-, pyroelectricity, magnetism and electro-optic effects.1,2 Perovskites were first applied as catalysts for oxidation of carbon monoxide in 1952–1953 by Parravano.1 For a typical ABO3 perovskite, the A-site is a larger rare earth and/or alkaline earth cation and the B-site is a smaller transition metal cation. Fine dispersion of “metal on oxide” catalyst can be obtained by reduction of the perovskite oxides.3 The advantages of perovskite materials could originate from the ability to accommodate a variety of ions of different valence with appropriate radius that meet the tolerance factor .3,4 The excellent tunable properties make perovskite potential candidate for catalysis.
.3,4 The excellent tunable properties make perovskite potential candidate for catalysis.
Recently, the perovskites with AA′BB′O3 structure have been used in catalysis since the substitution of A-site could influence the catalytic performance of B-site.5 The perovskites with special properties can be obtained by advisable tailoring. At present, the perovskite has been widely studied as the catalyst for NOx, CHx, and CO conversion.6
CO2 generated from fossil fuel combustion has led to environmental problem in the form of greenhouse effect and ozone depletion. For the sake of reducing the concentration of CO2 in atmosphere, various strategies have been implemented such as capture, storage, and utilization of carbon dioxide.7–10 An important means of carbon dioxide utilization is the hydrogenation reaction. Among the products for carbon dioxide hydrogenation, methanol is considered as the most valuable since it can be used as solvent, alternative fuel and raw material for synthesis of olefins, aromatics or gasoline that derived from traditional petrochemical processes.11,12
Many catalytic systems have been employed for methanol synthesis from CO2 hydrogenation among which Cu-based catalysts have been regarded as most effective.13 Different promoters have also been investigated such as Zn, Al, Mg, Mn, B, Zr, Y, Ce,14–16 which can improve the catalytic performance via controlling the reactivity of the active site Cu phase by determining texture, exposure of the active sites and interaction pattern with reagents, products and reaction intermediates.17,18
Our previous works have found that the Cu-based catalysts from perovskite precursors exhibit better methanol selectivity compared with the other Cu-based catalysts due to the appearance of Cuα+ in the structure.19 The study for CO hydrogenation and CO2 hydrogenation over LaMn1−xCuxO3 perovskite oxides suggested that when the copper amount that substituted manganese was less than or equal to 50%, the LaMnO3 could keep the perovskite structure and show preferable performance for methanol synthesis because of the interaction of Cu+ and Mn.20 However, little work had been conducted on the influence of the fourth metal elements doping for the La–Mn–Cu–O perovskite catalysts. Therefore, in the present work, the La–M–Mn–Cu–O (M = Ce, Mg, Y, Zn) perovskites were prepared and characterized to obtain a clear insight in the influences of the fourth elements doping on the structure and the catalytic performance. The methanol synthesis from CO2/H2 was chosen as the model reaction for the perovskite materials to investigate the influence of the structure changing on the catalytic performance.
2. Experimental
2.1. Catalysts preparation
The La–M–Mn–Cu–O (M = Ce, Mg, Y, Zn) perovskite-type oxides were prepared by sol–gel method using citric acid as complexing agent. The ratio for La, M, Mn, Cu is 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2:0.5:0.5. Adequate amounts of the precursor salts (La(NO3)3·nH2O; Mn(NO3)2, 50% solution; Y(NO3)3·6H2O; Cu(NO3)2·3H2O; Zn(NO3)2·6H2O; Ce(NO3)3·6H2O; Mg(NO3)2·6H2O) along with citric acid were dissolved in deionized water at a molar ratio of 2:1 (metal cations: citric acid). The solution was heated to 353 K to remove the water, and then enhanced the temperature to 423 K until ignition. The resulting powder were finally calcined under air at 673 K for 2 h and then at 1073 K for 4 h. The La–Mn–Cu–O catalyst and Mg, Y, Zn, Ce doping catalysts were then denoted as P, Mg–P, Y–P, Zn–P and Ce–P, respectively.
:0.2:0.5:0.5. Adequate amounts of the precursor salts (La(NO3)3·nH2O; Mn(NO3)2, 50% solution; Y(NO3)3·6H2O; Cu(NO3)2·3H2O; Zn(NO3)2·6H2O; Ce(NO3)3·6H2O; Mg(NO3)2·6H2O) along with citric acid were dissolved in deionized water at a molar ratio of 2:1 (metal cations: citric acid). The solution was heated to 353 K to remove the water, and then enhanced the temperature to 423 K until ignition. The resulting powder were finally calcined under air at 673 K for 2 h and then at 1073 K for 4 h. The La–Mn–Cu–O catalyst and Mg, Y, Zn, Ce doping catalysts were then denoted as P, Mg–P, Y–P, Zn–P and Ce–P, respectively.
2.2. Characterization of catalysts
X-ray diffraction (XRD) analysis of powered samples was performed using a PANalytica X'Pert Pro X-ray diffractometer with Cu Kα radiation in the range of 10° < 2θ < 90° and a scan step of 0.1° min−1.The surface area of samples was determined by N2 adsorption–desorption at liquid nitrogen temperature 77.30 K, using a Micromeritics Tristar 3000 instrument. Sample degassing was carried out at 473 K prior to acquiring the adsorption isotherm. The special areas were calculated from the isotherm according to the Brunauer–Emmett–Teller (BET) method.
The dispersion of Cu (DCu) and exposed Cu surface area (SCu) were determined by dissociative N2O adsorption and carried out on Micromeritics AutoChem 2920 instrument. The catalysts (0.15 g) were first reduced in 5% H2–Ar mixture (30 mL min−1) for 2 h at 603 K, and the amount of hydrogen consumption was denoted as X. Then, the reduced samples cooled to 338 K and isothermally purged with Ar for 30 min, after which the sample was exposed to N2O (85 mL min−1) for 1 h to ensure all the metallic copper change into cuprous oxide (N2O + Cu → N2 + Cu2O). The samples were then flushed with Ar to remove the N2O and cooled to room temperature. Finally, a pulse of pure H2 was passed over the catalyst at 603 K. The surface Cu2O were reduced in the pulse of pure H2, and the amount of consumed H2 was denoted as Y. The dispersion of Cu and exposed Cu surface area of the catalyst were calculated by the eqn (1) (ref. 21) and eqn (3) (ref. 22)
 | (1) |
| nCu = 2Y | (2) |
| SCu = (nCu × N)/(1.4 × 1019 × W)(m2 g−1) | (3) |
The elemental composition was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo iCAP 6300), 100 mg of the each catalyst was dissolved in 5% HCl solution and diluted. The reference solutions were prepared with the metal nitrates or standard metal oxides used in the catalyst preparation.
Investigations of the sample microstructure morphology were performed on a FETXL30 S-FEG scanning electron microscope (SEM) with an accelerating voltage of 10.0 kV.
X-ray photoelectron spectroscopy (XPS) analyses were performed over a Kratos XSAM800 spectrometer equipped with Al Kα radiation (12 kV × 15 mA, hν = 1486.6 eV) under ultrahigh vacuum (10−7 Pa). The binding energies were calibrated internally by adventitious carbon deposit C(1s) with Eb = 284.6 eV (accuracy within ±0.1 eV). Samples were treated under pure hydrogen at 603 K for 2 h in the pre-treatment chamber before transferred to the analysis chamber.
Temperature program reduction (TPR) measurements were carried out in order to determine the reducible species and performed in a U-tube quartz reactor. The samples (50 mg) were purged with Ar (30 mL min−1) at 423 K to remove physically adsorbed water and then reduced in the flow of 5 vol% H2 + Ar (30 mL min−1) at a heating rate of 5 K min−1 up to 873 K. Thermal conductivity detector (TCD) was used to monitor the consumption of H2.
The adsorption property of H2 for the studied sample was measured by H2 temperature-programmed desorption (H2-TPD). The catalyst was first reduced at 603 K and cooled to 318 K to saturate for 60 min in H2 flow of 30 mL min−1 for 2 h. Then the catalyst was flushed with Ar flow (40 mL min−1) to remove all physical adsorbed molecules. Afterward, the TPD experiment was started with a heating rate of 10 K min−1 under Ar flow (40 mL min−1), and the change of hydrogen signal was monitored by a TCD and quantitatively calibrated by H2 pulses.
The basicity of the catalyst was measured by CO2 temperature-programmed desorption (CO2-TPD) was performed in the same way as that of H2-TPD; the only difference was that the desorbed CO2 was detected by a BALZER mass spectrometer. CO2 peak area was quantitatively calibrated by injecting CO2 pulses.
2.3. Evaluation of catalysts
Activity measurements in the hydrogenation of CO2 were carried out in a continuous-flow, high-pressure, fixed-bed reactor. Catalyst (1.5 g, 40–60 mesh) diluted with quartz sand (60 mesh) was placed in a stainless steel tube reactor. Prior to reaction, the catalyst was reduced in pure H2 at a flow-rate of 80 mL min−1 under atmospheric pressure. The reduction temperature was programmed to increase from room temperature to 603 K at a heating rate of 1 K min−1 and maintained at 603 K for 8 h. The reactor was then cooled to room temperature. After reduction, the activities of the catalyst samples in CO2 hydrogenation process were determined under reaction conditions of 523 K, 5.0 MPa, n(H2):n(CO2) = 3:1, GHSV = 4000 h−1. The steady-state activity measurements were taken after at least 24 h on the stream. Products were quantitative analyzed with gas chromatograph equipped with a TCD, and the columns were TDX-01 for gas of H2, CO and CO2 and the Propake-Q for liquid of water and methanol. The CO2 conversion and the carbon-based selectivity for CH3OH, CO and CH4 were calculated by an internal normalization method. The space time yields (STYs) of CH3OH, which gave the amounts of CH3OH produced per gram catalyst per hour, were defined as the eqn (4):
 | (4) |
3. Results and discussion
3.1. Textural and structural properties
The X-ray diffraction patterns of the fresh and reduced perovskites are presented in Fig. 1a and b respectively. It can be seen that the LaMnO3 (JCPDS # 75-0440) are the main phase for all samples. The diffraction peak at about 2θ = 32.5° shift towards higher values with the addition of the fourth elements. According to the Bragg formula, nλ = 2dsinθ (where d denotes the crystalline plane distance for indices (hkl), λ is the X-ray wave-length and θ is the diffraction angle), the d value will decrease when the ions with large radii are substituted by those with small radii (1.36 Å for La3+, 1.34 Å for Ce3+, 1.08 Å for Y3+, 0.74 Å for Zn2+, 0.73 Å for Cu2+, 0.72 Å for Mg2+, 0.65 Å for Mn3+), which results in the shifts of the diffraction peaks to higher values.23,24 The result implies that the fourth elements have strong influences on La. Since the Mg2+ is slightly smaller than Cu2+, it might also have a complex effect on perovskite structure. For P sample, only LaMnO3 phase is observed which means that all the copper either penetrates into the perovskite structure or well dispersed on the catalyst.20 With the addition of other ions, small peaks at 2θ = 35.6° and 38.9° assigned to CuO (JCPDS # 89-5899) appear and the intensity of the diffraction peak of the LaMnO3 phase increase in the order of: Mg–P < Ce–P < Y–P < P < Zn–P. The strength of the CuO peak is in the order of: Y–P < Ce–P < Zn–P < Mg–P, which suggests the influence on the perovskite structure with the introduction of the fourth elements. When Mg is introduced, the strongest diffraction peak of CuO and the weakest diffraction peak of LaMnO3 are found which implies the stronger impact of Mg for the penetration of copper into the catalyst lattice as well as the perfection of LaMnO3 in comparison with other elements. As Mg has the similar radius with Cu, it could have effect on both A-site and B-site of the perovskite oxide. However, doping with Y in the sample has little influence i.e. the characters of Y3+ are almost the same with La3+ which then lead to little disturbance for the catalyst structure. With the doping of Zn, the strongest diffraction peak of LaMnO3 is observed which suggests that the perovskite structure tends to perfection due to the similar radius, valence state and electronegativity for Zn and Cu. In all diffraction patterns, no phases that ascribe to Mg, Y or Zn is observed while a new phase ascribed to CeO2 is found for the sample of Ce–P which demonstrates that it is difficult for all the Ce to enter the perovskite lattice which agrees with the conclusion by Weng et al.25
 | ||
| Fig. 1 XRD patterns of the calcined (a) and reduced (b) perovskite-type catalysts: (□) LaMnO3; (●) CuO; (♦) CeO2; (*) Cu. | ||
As shown in Fig. 1b, for all reduced samples, LaMnO3 phase is still the main phase while the CuO phase disappears and the Cu phase emerges which reveals that the reduction process does not destroy the perovskite structure. However, the perovskite structure has undergone some changes, e.g. the phase symmetry of all samples has changed except for P (Table 1). It is interesting that the reduction process leads to ordered LaMnO3 phase only for Mg–P. The transition may occur in several steps and the deviation from the cubic perovskite structure may proceed from a simple distortion of the cubic unit cell, or an enlargement of cubic unit cell, or a combination of both.3 Moreover, the less particles shrink for Mg doped sample may result from the electronic property of alkaline-earth metals.
| Samples | Phase symmetry | Size of LaMnO3 crystallites (Å) | Elemental compositiona (ICP-OES) | SBET (m2 g−1) | Dispersionb (%) | SCu (m2 g−1) | ||
|---|---|---|---|---|---|---|---|---|
| Calcined | Reduced | Calcined | Reduced | |||||
| a Subscripts came from ICP results.b Calculated from N2O dissociative adsorption. | ||||||||
| P | Cubic | Cubic | 418 | 290 | La0.84Mn0.51Cu0.50 | 6.5 | — | — |
| Mg–P | Orthorhombic | Cubic | 206 | 332 | La0.67Mg0.22Mn0.49Cu0.50 | 5.4 | 0.9 | 1.2 |
| Y–P | Cubic | Hexagonal | 378 | 316 | La0.67Y0.23Mn0.47Cu0.50 | 11.3 | 3.8 | 4.6 |
| Zn–P | Cubic | Orthorhombic | 467 | 462 | La0.67Zn0.18Mn0.50Cu0.50 | 4.1 | 0.7 | 0.9 |
| Ce–P | Cubic | Orthorhombic | 333 | 238 | La0.68Ce0.19Mn0.49Cu0.50 | 7.2 | — | — |
The physicochemical properties of the calcined perovskite-type catalysts are also summarized in Table 1. The Y–P and the Zn–P possesses the largest and the lowest specific surface area, respectively. Moreover, the exposed Cu surface area and the Cu dispersion are measured by N2O adsorption technique. The largest copper surface area is observed for Y–P, nevertheless, the copper surface area cannot be measured for both P and Ce–P. Since the surface copper may have strong influence on the activity for CO2 hydrogenation reaction,26 the lower copper surface area may not favorable for the conversion of CO2. The ICP results show that the real contents of the samples are similar to the nominal values. For all catalysts, the experimental lanthanum amount is lower than the theoretical value.
Fig. 2 shows the SEM images of the prepared catalysts. The results show that all samples present as irregular granules. Compared with other samples, the particle size is smaller for Y–P and P. Particle agglomeration is observed for Mg–P and Ce–P while not for Zn–P in spite of the large particles.
 | ||
| Fig. 2 SEM images of the calcined catalysts (A) P, (B) Mg–P, (C) Y–P, (D) Zn–P, (E) Ce–P. | ||
3.2. The XPS investigations
The reduced perovskite-type catalysts are analyzed by XPS, and the binding energies (BE) of La 3d5/2, Mn 2p and O 1s are presented in Table 2. The La 3d5/2 peak at around 834.3 eV is close to the value of pure lanthana at 834.4 eV, indicating that lanthanum ions are present in the trivalent form.27 However, with the addition of the fourth elements, the BE of La 3d5/2 shift towards lower values which implies the increasing of the electron cloud density around La ions. It may due to the fourth elements affect the transfer of the electrons of La to O since O has the highest electronegativity value among all elements.28| Binding energy (eV) | Relative surface concentration of metala (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Samples | La 3d5/2 (eV) | Mn 2p3/2 (eV) | O 1s (eV) | Oad/O2 | La (%) | Mn (%) | Cu (%) | M (%) |
| a Values in parentheses are nominal concentration normalized to the total metal content. | ||||||||
| P | 835.1 | 642.2 | 529.2 (50.5%) | 1.02 | 46.7 (50) | 22.2 (25) | 31.1 (25) | — |
| 531.7 (49.5%) | ||||||||
| Mg–P | 834.0 | 642.0 | 529.1 (51.5%) | 1.06 | 25.3 (40) | 28.7 (25) | 25.6 (25) | Mg: 20.4 (10) |
| 531.5 (48.5%) | ||||||||
| Y–P | 834.4 | 641.3 | 529.0 (54.0%) | 1.18 | 26.1 (40) | 28.9 (25) | 32.7 (25) | Y: 12.3 (10) |
| 531.4 (46.0%) | ||||||||
| Zn–P | 834.1 | 641.5 | 529.1 (51.2%) | 1.05 | 38.5 (40) | 31.5 (25) | 21.3 (25) | Zn: 8.7 (10) |
| 531.5 (48.8%) | ||||||||
| Ce–P | 834.3 | 641.8 | 529.2 (51.9%) | 1.08 | 28.3 (40) | 27.8 (25) | 31.4 (25) | Ce: 12.5 (10) |
| 532.2 (48.1%) | ||||||||
For O 1s, the lower binding energy at around 528.9–529.1 eV can be ascribed to the lattice oxygen (O2−)15,28 and the BE value at around 530.8–533.0 eV is assigned to the adsorbed oxygen species (Oad) in the surface which contains hydroxyl (OH−), carbonate species (CO32−) and molecular water. Doped with the fourth components, the binding energy decreases which indicates that there are more electrons around oxygen. It is likely that the fourth components transfer the electronic to the oxygen. Obviously, the intensity of the peaks is different from each other. The presence of surface adsorbed oxygen species suggests the formation of oxygen vacancies in the defected oxides.29 The increasing of the Oad/O2− ratio upon addition of the fourth elements indicates that the increasing of the amount of defect on perovskite oxide that is favorable for the activation of the catalyst. The binding energy of Mn 2p3/2 for MnO, Mn2O3 and MnO2 are located at 640.6, 641.9 and 642.2 eV respectively. As reported in other studies,30,31 the mean oxidation state of Mn ions at the surface layers is extremely difficult to detect by XPS. However, the previous reports suggested that the BE difference between Mn 2p3/2 and O 1s increase with about 0.6–0.7 eV for the change of the oxidation state between Mn3+ and Mn4+. In this study, as shown in Table 2, the BE difference is in the range of 112.3–113.0 eV which means a change of the Mn4+/Mn3+ ratio.32,33
Since the binding energy of the Cu 2p3/2 band in the metal (932.6 eV) and in Cu+ (932.4 eV) are almost same, they can be distinguished by different kinetic energy of the Auger Cu LMM line position in Cu0 (918.6 eV), Cu+ (916.7 eV) or in Cu2+ (917.9 eV).26,34 The Auger electron spectroscopies of Cu LMM of reduced samples are shown in Fig. 3. The profiles are deconvoluted into two peaks. It can be seen that the majority of the copper species exist as Cu+ for all samples except for the P sample. The predominance of Cu+ in P is in accordance with the report of Jia et al.20 and the results of XRD and N2O-adsorption mentioned above. The weak Cu0 peak could be the explanation for the immeasurable of exposed Cu0 in the N2O-adsorption results for Ce–P (Table 1).
 | ||
| Fig. 3 Cu LMM Auger electron spectroscopy of (a) P; (b) Mg–P; (c) Y–P; (d) Zn–P; (e) Ce–P samples after reduction. | ||
The surface compositions and the nominal concentration of the catalysts are also listed in Table 2. The enrichment of Cu and depletion of La and Mn are observed for P. However, the Mn enrichment is found with the doping of the fourth elements. As Y and Ce entered the perovskite structure, it may lead to further enrichment of Cu on the catalyst surface. For Zn–P, the lack of both Zn and Cu on the surface may indicate that more Zn and Cu enter the bulk of the sample. It is also found that Mg is greatly enriched on the surface of Mg–P.
Fig. 4 shows the binding energy curves of the fourth components. The peak at 49.4 eV is attributed to Mg–O binding,35 1021.8 eV is assigned to Zn–O binding.30 While the 156.8 eV and 158.6 eV may originate from the Y existed as 3+.36 For Ce–P, six peaks located at 882.3, 888.4, 898.1, 900.7, 907.2 and 916.2 eV can be ascribed to Ce4+ which is in accordance with the XRD result of the Ce–P.37
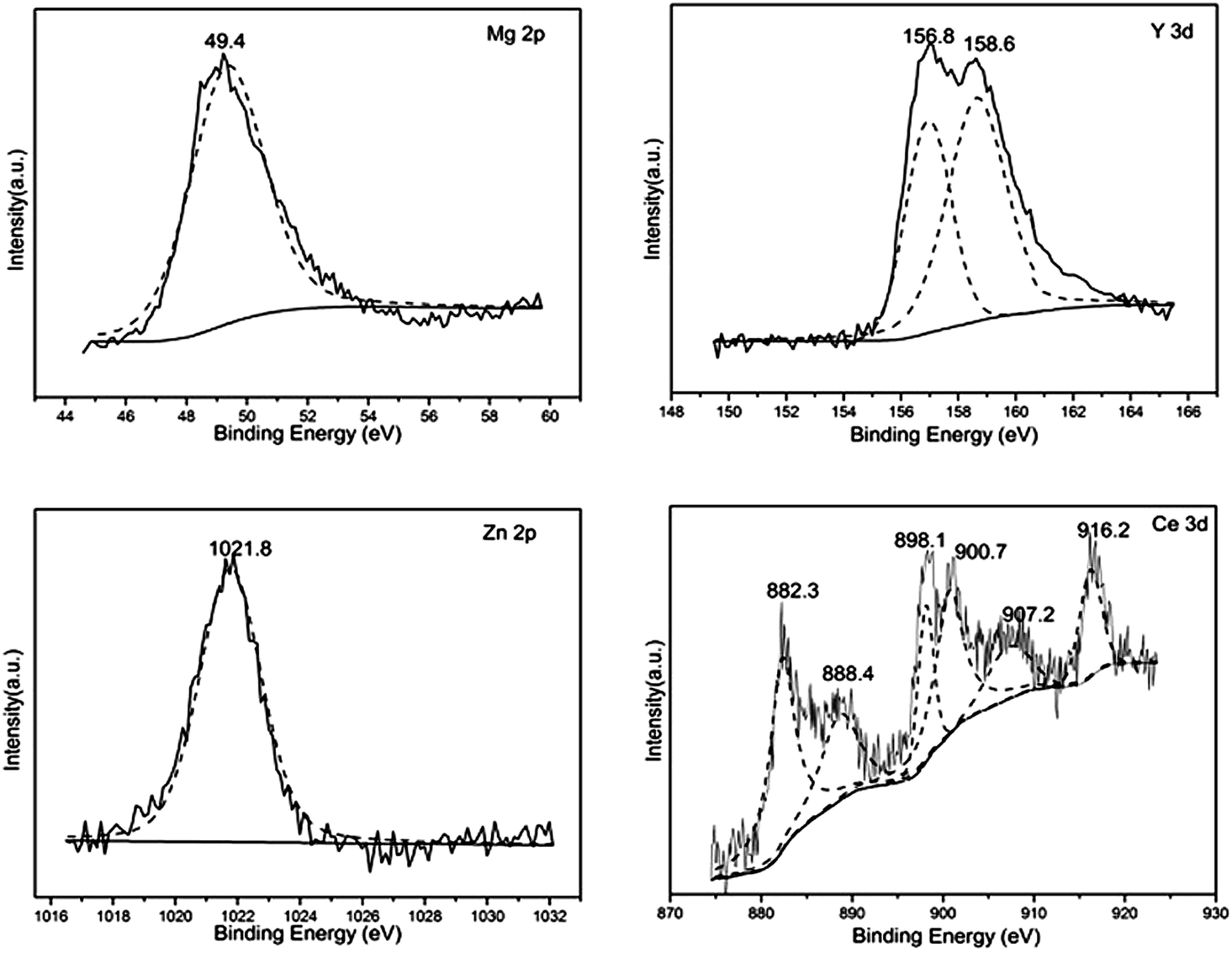 | ||
| Fig. 4 XPS results of Mg 2p, Y 3d, Zn 3d and Ce 3d. | ||
3.3. The reducibility of the prepared catalysts
The TPR profiles of all samples are shown in Fig. 5. It can be seen that the Y–P sample has two reduction peaks (denoted as peak α and peak β) while other samples present only one peak (denoted as peak β) which indicates that there is only one kind of copper spices in the P, Mg–P, Zn–P and Ce–P samples. The peak α at the lower temperature can be attributed to the reduction of highly dispersed surface copper species, whereas the peak at higher temperature is related to the reduction of the bulk-like copper species.38 Upon introduction of the fourth elements, the enrichment of the copper on the surface (Table 2) leads to lowered reduction temperature. Moreover, more Oad (Table 2) on the surface may also result in lowered reduction temperature which confirms the function of the defection in the perovskites. Y doping can increase the amount of the copper on the surface area which agrees with the N2O chemisorption. | ||
| Fig. 5 TPR profiles of the catalysts. | ||
The H2 consumption in the TPR experiment is listed in Table 3. The amount of H2 consumption is in the order of: Zn–P > Y–P > P > Mg–P > Ce–P, which indicates that there are more copper species that can be reduced for Zn–P and Y–P.
| Samples | H2 consumption (mmol g−1) | H2 desorption (μmol g−1) | ||||
|---|---|---|---|---|---|---|
| Peak α | Peak β | Total | H2-523a | H2-523b | ||
| a H2 desorption below 523 K.b H2 desorption below 523 K on per unit are (μmol g−1 m−2). | ||||||
| P | 9.47 | 48.2 | 165.2 | 213.4 | 8.7 | 1.34 |
| Mg–P | 9.06 | 50.6 | 68.3 | 118.9 | 18.9 | 3.50 |
| Y–P | 9.50 | 60.7 | 76.8 | 137.5 | 40.2 | 3.56 |
| Zn–P | 9.59 | 35.5 | 46.8 | 82.3 | 24.3 | 5.93 |
| Ce–P | 8.65 | 69.3 | 51.6 | 120.9 | 25.0 | 3.47 |
3.4. H2-TPD analysis
The H2-TPD profiles of the reduced catalysts are shown in Fig. 6. In order to get more insight into the H2-TPD results, the profiles are deconvoluted into two Gaussian peaks. According to the literature, the lower peak around 400 K can be assigned to the desorption of weakly molecularly adsorbed hydrogen on the Cu0 sites. However, such desorption peak are not observed because of less surface content of Cu0. The desorption peak (peak α) at lower temperature can be assigned to the desorption of dissociative hydrogen on the surface copper species, while the higher temperature peak (peak β) can be ascribed to desorption of strongly adsorbed dissociative hydrogen on the bulk copper or other metal oxides.39–41 It can be seen that the desorption peak of H2 at low temperature shifts toward even lower temperature with the introduction of the fourth components. However, the temperature of the peak β is higher than that for methanol synthesis, which means that these hydrogen might have little effect on the reaction. In addition, the desorption peak of the H2 presents as a broad peak also suggests the strong spillover in the perovskite catalysts. | ||
| Fig. 6 H2-TPD curves of the catalysts. | ||
The amount of H2 desorption over the pre-reduced materials is also listed in Table 3. With the introduction of the fourth elements, the total and high temperature desorption amount of hydrogen decreased. However, the desorption amount of H2 at low temperature increased except Zn–P. It implies that the doping of the fourth element is not favor the desorption of H2, especially under high temperature. Moreover, the H2 desorption on the unit surface area below 523 K (test temperature) increased for all the four components samples.
3.5. CO2-TPD analysis
Two desorption peaks (denoted as peak α and peak β) are observed for all samples (Fig. 7) which could be assigned to weak basic sites (around 400 K) and strong basic sites (around 600 K). The weak basic sites may be ascribed to the linear adsorption of CO2 while the strong basic sites may result from the bridge adsorption of CO2.41 With the introduction of the fourth components, the peak α shifts to higher temperature which indicates the increasing of the strength of the weak basic sites. The strength for the weak basic sites of the catalysts increases in the order of: P < Ce–P < Y–P < Mg–P < Zn–P. However, the peak β shifts to lower temperature which reveals that the addition of the fourth elements decrease the basicity of the strong basic sites of the samples. | ||
| Fig. 7 CO2-TPD curves of the catalysts. | ||
Doping with the fourth elements leads to the change of the amount of the basic sites. The quantitative analysis for the CO2-TPD based on the relative area of the profiles is listed in Table 4, in which the P sample is assigned as 1.00. For Mg–P, the amount of all basic sites increases due to the alkalinity of Mg. Although the amount of total basic sites and strong basic sites for Y–P increases, the amount of weak basic sites decreases. Moreover, the amount of the weak basic sites increases for Zn–P and Ce–P samples despite the decrease of the amount of the strong basic sites and total basic sites.
| Samples | Adsorption type and distribution based in CO2-TPD dataa | ||
|---|---|---|---|
| Peak α | Peak β | Total | |
| a The amount of basicity of P is assigned as 1.00 to compare with other samples and the values in parentheses are the desorption temperature (K). | |||
| P | 1.00 (383) | 1.00 (614) | 1.00 |
| Mg–P | 1.30 (396) | 1.06 (583) | 1.11 |
| Y–P | 0.87 (387) | 1.67 (595) | 1.53 |
| Zn–P | 1.56 (401) | 0.85 (596) | 0.97 |
| Ce–P | 1.22 (385) | 0.69 (581) | 0.78 |
3.6. Catalytic performance
The catalytic performances of the catalysts for CO2 hydrogenation to methanol are summarized in Table 5. The main product is methanol and the by-products are CO and H2O derived from reverse water gas shift (RWGS) reaction.34,38,42 It is found that the CO2 conversion and methanol selectivity are improved for the doped catalysts. With the introduction of Zn, both the CO2 conversion and the methanol selectivity increased greatly which might due to the fact that CO2 could be activate on the site of Cu+–O–Zn.43,44 However, the addition of Ce leads to a slight improvement for the catalytic performance. The relationship between the CO2 conversion and the amount of H2 desorption on unit surface area below 523 K (Table 3) is shown in Fig. 8. It can be seen that the more H2 desorbed on the unit area, the more CO2 conversion. Lower CO2 conversion may result from lower copper content as well as lower surface area of copper in the system. The results of Fig. 9 indicate that the strength of the weak basic sites play an important role for methanol selectivity. The formate theory may explain the relationship between the catalytic performance and the H2 desorption as well as the strength of the weakly basic sites in which the formaldehyde is the intermediate species and the basic sites is the key factor for methanol selectivity.43| Samples | CO2 conversion (%) | Selectivity (C mol%) | ||
|---|---|---|---|---|
| CH3OH | CO | CH4 | ||
| a Reaction conditions: n(H2)/n(CO2) = 3:1, T = 523 K, P = 5.0 MPa, GHSV = 4000 h−1. |
||||
| P | 1.8 | 0.7 | 93.4 | 5.9 |
| Mg–P | 2.8 | 23.7 | 68.1 | 6.5 |
| Y–P | 4.6 | 14.5 | 82.6 | 2.9 |
| Zn–P | 6.1 | 51.0 | 46.4 | 2.7 |
| Ce–P | 2.0 | 5.0 | 85.9 | 9.2 |
 | ||
| Fig. 8 The relationship between the CO2 conversion and the amount of H2 desorbed on unit surface area below 523 K. | ||
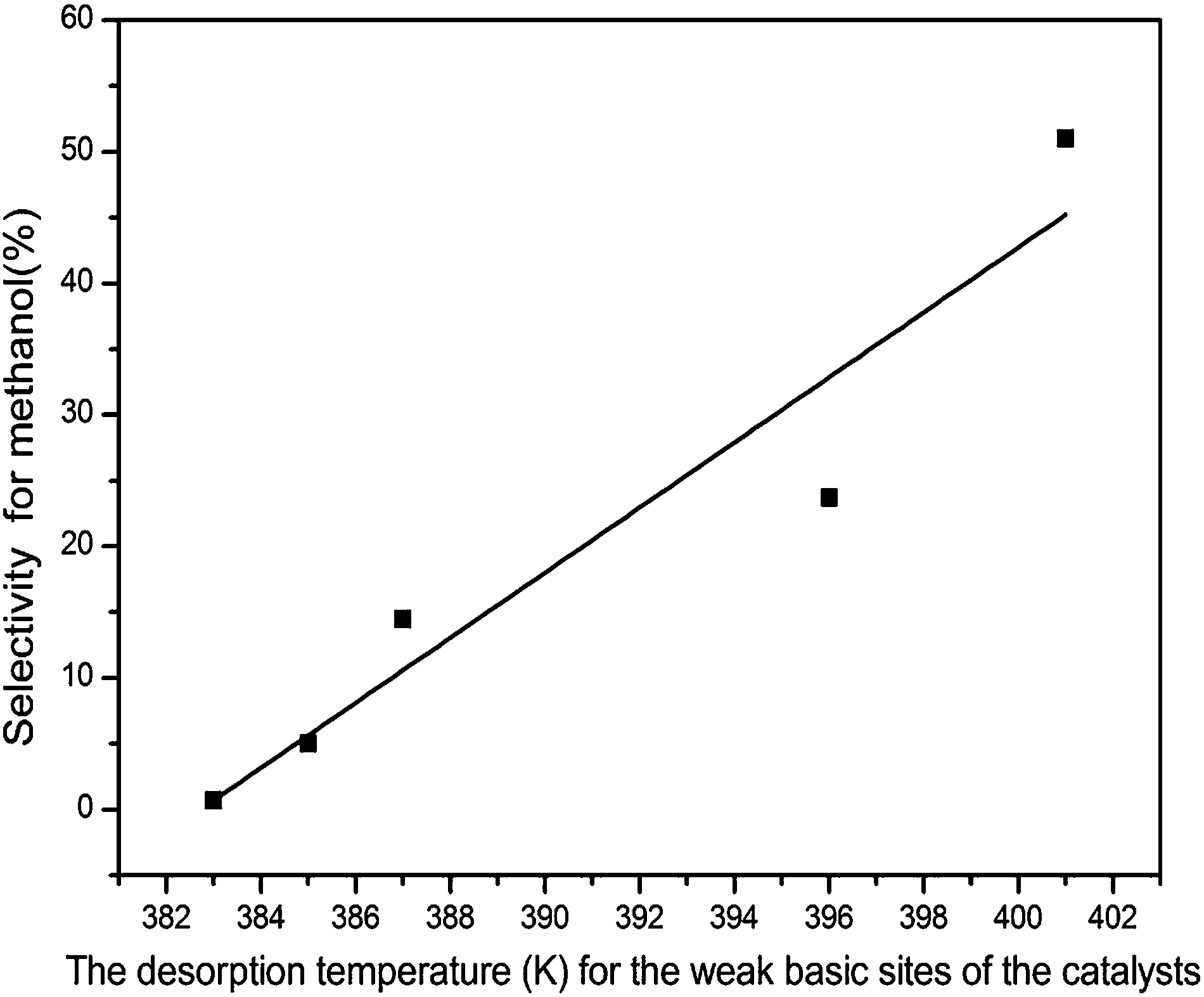 | ||
| Fig. 9 The relationship between the selectivity for methanol and the strength of the weak basic of the catalysts. | ||
Previous studies have demonstrated that two active centers were involved in the catalytic process of CO2 hydrogenation for Cu-based catalysts which undergo the formate intermediate process,26,45 i.e. CO2 was adsorbed on the surface of the metal oxides such as ZnO and ZrO2 while the H2 was adsorbed on the Cu sites. Then the activated species participated in the reaction to produce the aim products.43,46,47 During the reaction, either methanol or by-product CO was produced from the intermediate, formate species, and the strength of the basic sites decided the products selectivity. With the special perovskite structure, in our study, CO2 could be adsorbed on the basic sites (perovskite oxide carriers) to form activated CO*2 and H2 may be adsorbed and activated on the Cu sites resulting from the reduction of extra-perovskite and intra-perovskite copper species. The methanol selectivity is only related to the strength of the weak basic sites. The hydrogen supplied from Cu sites may spillover to the basic sites and react with the CO2* to form the formate intermediate. The formate species formed on the weak basic sites may lead to methanol production, while the intermediate formed on other basic sites may produce CO. The possible reaction mechanism for CO2 hydrogenation to methanol over the perovskites catalysts can be described in Scheme 1.
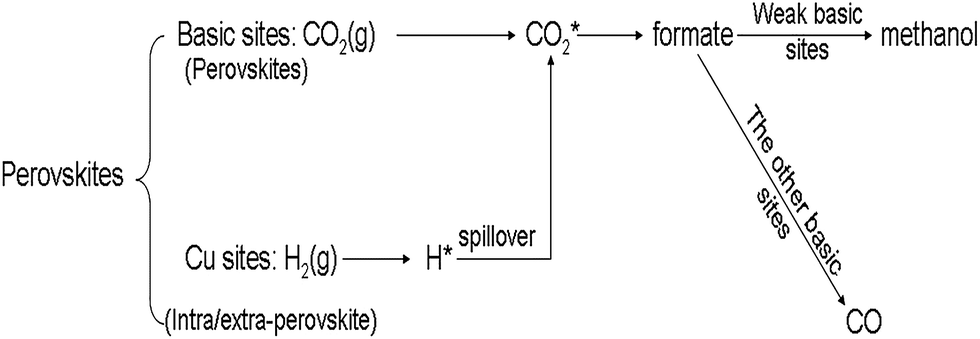 | ||
| Scheme 1 Proposed reaction mechanism of CO2 hydrogenation to methanol over the perovskite catalysts. | ||
4. Conclusions
Doped La–M–Mn–Cu–O (M = Ce, Mg, Y, Zn) catalysts derived from perovskite-type precursors were prepared by sol–gel method. Introduction of the fourth elements leads to the separation of copper out of the LaMnO3 perovskite lattice and thus produces more oxygen vacancies. Lower reduction temperature and low temperature adsorption properties are obtained with the increasing of defects for the doped samples. For the reaction of CO2 hydrogenation to methanol, the CO2 conversion increases with increasing of the amount of absorbed H2 on the unit area under 523 K, while the methanol selectivity increases upon increasing of the strength of the weak basic sites. Both the H2 adsorption amounts on the unit area and weak basic site strength are enhanced considerably as a result of the introduction of Zn such that improved the catalytic performance. Firstly, the Zn doped sample showed the largest H2 consumption in the TPR result which indicated the existence of the most amount of reducible copper (the active site for the reaction) in the sample. Secondly, the Zn doped sample possessed the maximum amount of H2 desorption on the unit surface area under 523 K. Thirdly, the samples possessed more weakly basic sites which favored the methanol selectivity. The Ce–P shows an iso-conversion compared with P due to the slight improvement of the H2 adsorption. However, with the slight enhancement of the weak basic sites, the Ce–P showed almost five times more selectivity towards methanol which implies that it is probably much easier to improve the methanol selectivity for the perovskite catalytic system.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (21343012), the National Key Technology Research and Development Program of the Ministry of Science and Technology (2013BAC11B00) and “Strategic Priority Research Program-Climate Change: Carbon Budget and Related Issues” of the Chinese Academy of Sciences, Grant no. XDA05010109, XDA05010110, XDA05010204.Notes and references
- L. G. Tejuca, J. L. G. Fierro and J. M. D. Tascón, Adv. Catal., 1989, 36, 237–328 CAS.
- X. F. Sun, S. Komiya and Y. Ando, Phys. C, 2003, 388–389, 355–356 CrossRef CAS.
- M. A. Pẽna and J. L. G. Fierro, Chem. Soc. Rev., 2001, 1981–2017 CrossRef PubMed.
- C. D. Clive, C. Roger and M. J. Hampden-Smith, Chem. Soc. Rev., 1993, 93, 1205–1241 CrossRef.
- J. L. G. Fierro, M. L. Rojas, L. G. Tejuca and A. T. Bell, J. Catal., 1990, 124, 41–51 CrossRef.
- J. Zhang, X. Weng, Z. Wu, Y. Liu and H. Wang, Appl. Catal., B, 2012, 126, 231–238 CrossRef CAS PubMed.
- B. Li, Y. Duan, D. Luebke and B. Morreale, Appl. Energy, 2013, 102, 1439–1447 CrossRef CAS PubMed.
- R. Steeneveldt, B. Berger and T. A. Torp, Chem. Eng. Res. Des., 2006, 84, 739–763 CrossRef CAS.
- K. M. Yu, I. Curcic, J. Gabriel and S. C. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- K. Damen, M. van Troost, A. Faaij and W. Turkenburg, Prog. Energy Combust. Sci., 2006, 32, 215–246 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- J. Słoczynski, R. Grabowski, P. Olszewski, A. Kozlowska, J. Stoch, M. Lachowska and J. Skrzypek, Appl. Catal., A, 2006, 310, 127–137 CrossRef PubMed.
- J. Słoczyński, R. Grabowski, A. Kozłowska, P. Olszewski, M. Lachowska, J. Skrzypek and J. Stoch, Appl. Catal., A, 2003, 249, 129–138 CrossRef.
- F. Meshkini, M. Taghizadeh and M. Bahmani, Fuel, 2010, 89, 170–175 CrossRef CAS PubMed.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, Catal. Today, 2013, 210, 39–46 CrossRef CAS PubMed.
- F. Arena, G. Mezzatesta, G. Zafarana, G. Trunfio, F. Frusteri and L. Spadaro, J. Catal., 2013, 300, 141–151 CrossRef CAS PubMed.
- H. J. Zhan, F. Li, P. Gao, N. Zhao, F. K. Xiao, W. Wei, L. S. Zhong and Y. H. Sun, J. Power Sources, 2014, 251, 113–121 CrossRef CAS PubMed.
- L. S. Jia, J. Gao, W. P. Fang and Q. B. Li, J. Rare Earths, 2010, 28, 747–751 CrossRef CAS.
- Z. Yuan, L. Wang, J. Wang, S. Xia, P. Chen, Z. Hou and X. Zheng, Appl. Catal., B, 2011, 101, 431–440 CrossRef CAS PubMed.
- R. Yang, X. Yu, Y. Zhang, W. Li and N. Tsubaki, Fuel, 2008, 87, 443–450 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- J. P. Zou, L. Z. Zhang, S. L. Luo, L. H. Leng, X. B. Luo, M. J. Zhang, Y. Luo and G. C. Guo, Int. J. Hydrogen Energy, 2012, 37, 17068–17077 CrossRef CAS PubMed.
- D. Weng, H. Zhao, X. Wu, L. Xu and M. Shen, Mater. Sci. Eng., A, 2003, 361, 173–178 CrossRef.
- P. Gao, F. Li, H. J. Zhan, N. Zhao, F. K. Xiao, W. Wei, L. S. Zhong, H. Wang and Y. H. Sun, J. Catal., 2013, 298, 51–60 CrossRef CAS PubMed.
- N. Merino, B. Barbero, P. Grange and L. Cadus, J. Catal., 2005, 231, 232–244 CrossRef CAS PubMed.
- K. Sutthiumporn and S. Kawi, Int. J. Hydrogen Energy, 2011, 36, 14435–14446 CrossRef CAS PubMed.
- Z. Li, M. Meng, Y. Zha, F. Dai, T. Hu, Y. Xie and J. Zhang, Appl. Catal., B, 2012, 121–122, 65–74 CrossRef CAS.
- F. Rubio-Marcos, A. Quesada, M. A. García, M. A. Bañares, J. L. G. Fierro, M. S. Martín-Gonzalez, J. L. Costa-Krämer and J. F. Fernández, J. Solid State Chem., 2009, 182, 1211–1216 CrossRef CAS PubMed.
- Y. H. Kenji Tabata and E. Suzuki, Appl. Catal., A, 1998, 170, 245–254 CrossRef.
- N. H. Batis, P. Delichere and H. Batis, Appl. Catal., A, 2005, 282, 173–180 CrossRef CAS PubMed.
- H. Najjar and H. Batis, Appl. Catal., A, 2010, 383, 192–201 CrossRef CAS PubMed.
- P. Gao, F. Li, N. Zhao, F. K. Xiao, W. Wei, L. S. Zhong and Y. Sun, Catal. Sci. Technol., 2012, 2, 1447–1454 CAS.
- Y. Aykut, G. N. Parsons, B. Pourdeyhimi and S. A. Khan, Langmuir, 2013, 29, 4159–4166 CrossRef CAS PubMed.
- L. Dusoulier, R. Cloots, B. Vertruyen, J. L. Garcia-Fierro, R. Moreno and B. Ferrari, Mater. Chem. Phys., 2009, 116, 368–375 CrossRef CAS PubMed.
- A. F. Lucrédio, G. T. Filho and E. M. Assaf, Appl. Surf. Sci., 2009, 255, 5851–5856 CrossRef PubMed.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, J. Mol. Catal. A: Chem., 2011, 345, 60–68 CrossRef CAS PubMed.
- X. Dong, H. B. Zhang, G. D. Lin, Y. Z. Yuan and K. R. Tsai, Catal. Lett., 2003, 85, 3–4 CrossRef.
- F. Arena, G. Italiano, K. Barbera, G. Bonura, L. Spadaro and F. Frusteri, Catal. Today, 2009, 143, 80–85 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang and S. Chen, Appl. Catal., A, 2012, 415–416, 118–123 CrossRef CAS PubMed.
- P. Gao, F. Li, F. K. Xiao, N. Zhao, W. Wei, L. S. Zhong and Y. H. Sun, Catal. Today, 2012, 194, 9–15 CrossRef CAS PubMed.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS PubMed.
- B. J. A. Miller, M. A. Martin-luengo, M. S. W. Vong, Y. Wang, V. A. Self, S. M. Chapman and P. A. Sermon, J. Mater. Chem., 1997, 7, 2155–2160 RSC.
- E. E. Ortrlli, J. M. Weigel and A. Wokaun, Catal. Lett., 1998, 54, 41–48 CrossRef.
- C. Yang, Z. Ma, N. Zhao, W. Wei, T. Hu and Y. Sun, Catal. Today, 2006, 115, 222–227 CrossRef CAS PubMed.
- G. Q. L. Xin-Mei Liu, Z.-F. Yan and J. Beltramini, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |