
Detection of dopamine on a poly(metanilic acid) decorated two-dimensional gold cavity array electrode†
Abstract
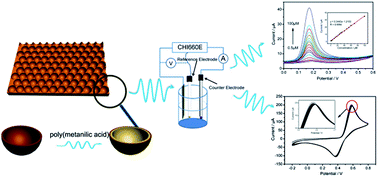
A shape-controllable, highly ordered, two-dimensional gold cavity array (GCA) electrode was prepared by electrodeposition using a closely packed monolayer of 1.2 μm-diameter polystyrene spheres as a template and was characterized by FESEM and XRD. A significant enhancement for the electrooxidation of dopamine at this nanostructured electrode was found due to the increased amount of the active surface area. By electropolymerizing a poly(metanilic acid) thin film on its surface, the enhanced electrochemical properties of the GCA electrode were maintained, and the antifouling property was improved. The SWV technique was used for the trace determination of DA, and the dependence of current vs. concentration was linear from 0.2 to 100 μM with a regression coefficient of 0.9988, and the detection limit of DA was ∼0.08 μM. Furthermore, the signals of DA and UA can be well distinguished at this poly(metanilic acid) modified GCA electrode. The proposed method was applied to the selective and precise analysis of DA in commercial injections.
 Please wait while we load your content...
Please wait while we load your content...

