
A hollow porous magnetic nanocarrier for efficient near-infrared light- and pH-controlled drug release†
Weidong Jia,
Najun Li*ac,
Dongyun Chena,
Yang Jiaob,
Qingfeng Xuac and
Jianmei Lu*ac
aJiangsu Key Laboratory of Advanced Functional Polymer Design and Application, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, China. E-mail: linajun@suda.edu.cn; lujm@suda.edu.cn; Fax: +86-512-6588-0367; Tel: +86-512-6588-0367
bSchool of Radiation Medicine and Protection, Medical College of Soochow University, Suzhou, Jiangsu 215123, China
cState Key Laboratory of Treatments and Recycling for Organic Effluents by Adsorption in Petroleum and Chemical Industry, Suzhou, 215123, China
First published on 29th September 2014
Abstract
A multi-functional core–shell nanocarrier was successfully prepared for near-infrared light- and pH-controlled drug release as well as magnetic resonance imaging (MRI) and fluorescence imaging. On the one hand, the hollow porous Fe3O4 (HPFe3O4, ∼20 nm) which could be etched under acidic conditions inside the cancer cells was prepared as the “core” to load anti-cancer drugs. On the other hand, the targeting NIR light-responsive copolymer (DDACMM-PEG-FA) was synthesized by polymerization of coumarin-containing monomer (DDACMM), poly(ethylene glycol) (PEG) methyl ether methacrylate and N-hydroxysuccinimide (NHS) and then modified by folic acid (FA). The core–shell nanocarriers (HPFe3O4@DDACMM-PEG-FA) were obtained by coating the amphiphilic copolymers onto the hollow porous “core”. Since the copolymer could be disrupted under the irradiation of NIR light laser (800 nm) via a two-photon absorption process, the pre-loaded drugs (∼65–80%) could be released from the nanocarriers. More importantly, the subacid environment in the tumour could further etch the boundary area of uncovered HPFe3O4, which further improved the efficiency of the drug release (about 20% increase in 24 h). The in vitro experiments indicated that the nanocarriers were biocompatible and could easily target the tumour cells that over-expressed folic acid receptor (FR(+)) and release the pre-loaded drugs successfully. In addition, because of the superparamagnetism of HPFe3O4 and the fluorescence of the polymer, the MRI and cell fluorescence imaging could be used to track the process of drug delivery.
Introduction
Over the last decade, significant progress has been made in designing and synthesising various amphiphilic polymers for surface modification of inorganic nanoparticles for drug delivery and release.1–3 These polymers are triggered by external stimuli such as enzymes,4 temperature5 and pH.6–8 There has recently been growing interest in light-responsive polymer because it could control over the moment and the location of drug release to provide a greater selectivity.9–14 Near-infrared (NIR) light is a kind of long-wavelength light which shows less damage to irradiated area with a greater penetration depth of the skin and high spatial precision.15–20 Due to these advantage, NIR light is more potential in clinic therapy than ultraviolet (UV) light.In our last research, we combined hollow mesoporous silica (HMS) with a NIR-light responsive polymer, the HMS played as a stable core to load drug and the polymer was a trigger for drug release.21 Recently, magnetic nanoparticles have been heavily pursued as versatile carriers for diagnostic and therapeutic applications.22,23 These nanoparticles are supermagnetic and are excellent contrast agent for magnetic resonance imaging (MRI), such as superparamagnetic iron oxide nanoparticles (SPIONPs).24–29 However, most of SPIONPs are solid sphere, whose non-porous structure results in low drug loading capacity. So the hollow porous SPIONPs with large pore volume are into the line of sight of researchers.30 Furthermore, the pore size could be controlled even readily by acid etching, because SPIONPs in the boundary area were more reactive and tend to be etched away first, then leaving the open pores on the shell structure. So that offers a way of controlling drug release in the slightly acid environment of the tumour.31–33
Here we fabricated a novel NIR light- and pH-responsive nanocarrier for controlled drug release and cell imaging by combining hollow porous Fe3O4 (HPFe3O4) and NIR light-responsive polymer via self-assembly (Scheme 1). To improve the biocompatibility of the nanocarrier, we used polyethylene glycol (PEG) as the hydrophilic segment.34 The multifunctional polymer was synthesized by free radical polymerization of [7-(didodecylamino)coumarin-4-yl] methyl methacrylate (DDACMM) and poly(ethylene glycol) methyl ether methacrylate (PEG) and N-hydroxysuccinimide (NHS), and then successfully conjugated with folic acid for selective cancer targeting. We easily wrapped amphipathic copolymers around the HPFe3O4 through the strength of van der Waals forces via a simple self-assembly strategy. The as-synthesized nanocarrier had excellent biocompatibility and NIR light responsivity. Doxorubicin (DOX, a widely used anticancer drug) was encapsulated in the above polymer-coated HPFe3O4 in advance. The resulting nanocarrier was specialized due to the NIR light-responsive layer, which could release pre-loaded DOX upon the photocleavage of the chromophores in the polymer layer. What is more important, the subacid environment in tumour cells could further etch the boundary area of uncovered HPFe3O4 and improved the efficiency of the drug release. Since the superparamagnetism of HPFe3O4 and the fluorescence of polymer, the MRI and cell fluorescent imaging were also investigated.
 | ||
| Scheme 1 Schematic depiction of the fabrication of HPFe3O4@DDACMM-PEG-FA and controlled release upon NIR light exposure. | ||
Experiment section
Materials
Azobisisobutyronitrile (AIBN), iron pentacarbonyl and poly(ethylene glycol) methyl ether methacrylate (PEG, Mn = 950) were purchased from Aldrich. Folate, 1-octadecene, oleylamine, oleic acid, dibenzyl ether, trimethyl amine N-oxide and N-succinimidyl acrylate (NSA) were from TCI Shanghai. The cyclohexanone was refined by distillation to remove polymerization inhibitor. Other reagents were commercially available and used as received. All the reactions were carried out at ambient conditions unless otherwise stated.Synthesis of hollow porous Fe3O4 (HPFe3O4)
The synthetic procedure was achieved according to the literature as reported elsewhere.31–35Synthesis of NIR-light responsive polymer (DDACMM-PEG-FA)
A round-bottom flask was charged with [7-(didodecylamino)coumarin-4-yl] methyl methacrylate (DDACMM) (168 mg, 0.28 mmol), PEG (1.6 g, 1.69 mmol), NSA (23 mg, 0.14 mmol), AIBN (3.4 mg, 0.021 mmol) and 3 mL refined cyclohexanone under N2 atmosphere. The monomer DDACMM was synthesized by our last research.21 The reaction mixture was degassed by three freeze–pump–thaw cycles, back filled with N2. After 8 h polymerization at 70 °C, the mixture was cooled to room temperature and then precipitated from petroleum ether, filtered and dried in vacuum. 1H NMR (CDCl3, 400 MHz), δ (ppm): 4.08 (COOCH2CH2), 3.89–3.14 (CH2OCH2), 1.24 (CH2), 0.85 (CH3).168 mg DDACMM-PEG and 4.0 mg folate were dissolved in 5 mL dimethyl sulfoxide. After being stirred for 48 h at room temperature, the mixture was precipitated in 40% acetone in diethyl ether. The filtrate was concentrated and stored in the dark at 4 °C. 1H NMR (d6-DMSO, 400 MHz), δ (ppm): 8.64–6.65 (folic acid), 4.00 (COOCH2CH2), 3.85–2.99 (CH2OCH2), 1.24 (CH2), 0.84 (CH3).
Self-assembly of DDACMM-PEG-FA with HPFe3O4 (HPFe3O4@DDACMM-PEG-FA)
HPFe3O4 (5 mg) and DDACMM-PEG-FA (60 mg) were dissolved in tetrahydrofuran (1 mL) and ultrasonic dispersed for 1 h. Then, 5 mL of distilled water was added to this solution with vigorous shaking. The mixture was kept stirring for 24 h at room temperature to evaporate tetrahydrofuran. After that, the product was separated by centrifugation and washed with distilled water several times to remove the unbound copolymer. The obtained product was denoted as HPFe3O4@DDACMM-PEG-FA.Hydrolysis test of DDACMM-PEG-FA
In order to analyze the hydrolysis rate of DDACMM-PEG-FA, we used NIR light and UV light to irradiate the same sample of DDACMM-PEG-FA water solution in quartz plate. And the hydrolysis value was determined by UV-Vis spectrum at λ = 385 nm. First, the quartz plate was filled of water solution of the polymer, and we measured the absorption value every once in a while after light irradiation.Drug loading and release
To evaluate the drug loading and release efficiency, doxorubicin (DOX) was used as a model anticancer agent. The DOX solution (40 μL, 5 mg mL−1) was diluted with water (1 mL), then HPFe3O4 (4.0 mg, 2.0 mg, 0.4 mg) was added to this solution. After stirring for 24 h in the darkness at room temperature, the HPFe3O4 with adsorbed DOX was separated by centrifugation, and washed with water for 3 times. And then the DOX-loaded HPFe3O4 was self-assembled with polymer in water. DOX-loaded HPFe3O4 (5 mg) and DDACMM-PEG-FA (60 mg) were dissolved in tetrahydrofuran (1 mL) and then 5 mL of distilled water was added to this solution with vigorous shaking. The mixture was kept stirring for 24 h at room temperature to evaporate tetrahydrofuran. After that, the product was separated by centrifugation and washed with distilled water several times to remove the unbound copolymer. The obtained product was denoted as DOX-loaded HPFe3O4@DDACMM-PEG-FA.The drug release test was performed by suspending the DOX-loaded nanoparticles in phosphate buffer solution (PBS) with shaking in a water bath maintaining the temperature at 37 °C under continuous NIR light exposure. To determine the release amount at any given time, 100 μL of the solution was withdrawn after centrifugation. The concentration of the DOX solution was determined using a fluorescence spectrophotometer at λex = 475 nm and λem = 592 nm and a standard plot had been prepared under identical conditions. The experiments were conducted in triplicate and the results are average data with standard deviations.
In vitro experiments
To test the cytotoxicity of various materials with and without NIR irradiation, KB cells were seeded in a 96-well plate at a density of ∼200 cells per well and cultured in 5% CO2 at 37 °C for 24 h. Then, different NPs were added to the medium. For materials without NIR irradiation, the cells were incubated in 5% CO2 at 37 °C for 24 h. For materials with NIR irradiation, the NPs were added, followed by exposing to NIR light immediately. Then, the cells were further incubated for 24 h in the dark. At the end of the incubation, the medium was removed, and 100 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (diluted in a culture media with a final concentration of 0.5 mg mL−1) was added and incubated for another 4 h. The medium was then replaced with 100 μL of dimethyl sulfoxide (DMSO) per well, and the absorbance was monitored using a microplate reader (Bio-TekELx800) at the wavelength of 490 nm. The cytotoxicity was expressed as the percentage of cell viability compared to untreated control cells.
Cellular uptake of the HPFe3O4@DDACMM-PEG-FA
KB cells were seeded in 96-well plates (1.3 × 104 per well) and incubated overnight at 37 °C in a humidified incubator. The dispersion of the HPFe3O4@DDACMM-PEG-FA sample was prepared in RPMI-1640 medium. Cells were cultured with the HPFe3O4@DDACMM-PEG-FA solution for a certain time and observed using confocal laser fluorescence microscopy (Olympus, FV 1000) after washing three times with PBS.Flow cytometry
Human KB cells were incubated as monolayer in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum. The medium was then replaced with a freshly prepared medium containing HPFe3O4@DDACMM-PEG-FA (with FA targeting groups) and HPFe3O4-DDACMM-PEG (without FA targeting groups). The cells were incubated for the desire time. After that, the suspensions were centrifuged and washed with cold PBS solution, and then resuspended in PBS. Flow cytometry was performed on BD FACS Calibur.Characterization
Gel permeation chromatography (GPC) analysis was carried out on a Waters 1515 pump and a differential refractometer, DMF was used as a mobile phase at a flow rate of 1.0 mL min−1. 1H NMR spectra were measured by an INOVA 400 MHz NMR instrument. TEM images were obtained using a TecnaiG220 electron microscope at an acceleration voltage of 200 kV. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) analyses were used to determine the surface area. Confocal laser scanning microscopy (CLSM) images were observed by a confocal laser scanning microscope (Olympus, FV 1000). The UV-Vis absorption spectra were measured on a TU-1901 spectrophotometer. Emission and excitation spectra were obtained using Edinburgh-920 fluorescence spectra photometer. Powder X-ray diffraction (XRD) experiments were performed on an X'Pert-Pro MPD X-ray diffractometer.Light exposure
The femtosecond pulse NIR photons were generated from a Coherent Vitesse Ti:Sapphire laser at 800 nm with a repetition rate of 1 kHz. The output power of the laser was controlled at 1.3–1.5 W. The continuous-wave diode NIR laser (808 nm, 4.0 W) was generated from LEO Photonics Co., Ltd. NIR beam whose spot size was approximately 2 mm in diameter was focused onto a 1 cm thick cuvette. For UV (365 nm) irradiation, a 125 W high-pressure mercury lamp from Philips was used.MRI experiments
MRI experiments were carried out with a 0.5 tesla (T) superconducting unit (GE Vectra, International General Electric, Slough, UK). HPFe3O4@DDACMM-PEG-FA was loaded into eppendorf tubes for imaging. Multisection T2-weighted fast spin-echo sequences were performed in at least two orthogonal planes to obtain MR phantom images. MRI signal intensity was calculated using the in-built software.Results and discussion
Synthesis of DDACMM-PEG-FA and HPFe3O4
The amphiliphilic photo-responsive polymer DDACMM-PEG-FA was synthesized by free radical polymerization as Scheme 2. The 1H NMR spectrum of DDACMM-PEG-FA (Fig. S2†) shows the successful conjugation of FA. The number-average molecular weight (Mn), weight-average molecular weight (Mw) and polydispersity index (PDI) of the polymers are shown in Table S1,† and the molecular weight increasing shows the successful conjugation of folate (∼28.3%). A typical 1H NMR spectrum of the copolymer DDACMM-PEG in CDCl3 is shown in Fig. S3a.† Degradation was observed by comparing the spectra of the copolymer before and after light exposure. After light exposure for several hours, the specific chemical shifts appear at δ = 8.058, 8.082, 4.407 and 4.318 ppm, which corresponds to the carboxylic acid group and coumarin hydroxyl group, indicates the obvious photo-degradation process of the polymer chain under light exposure (Fig. S3b†).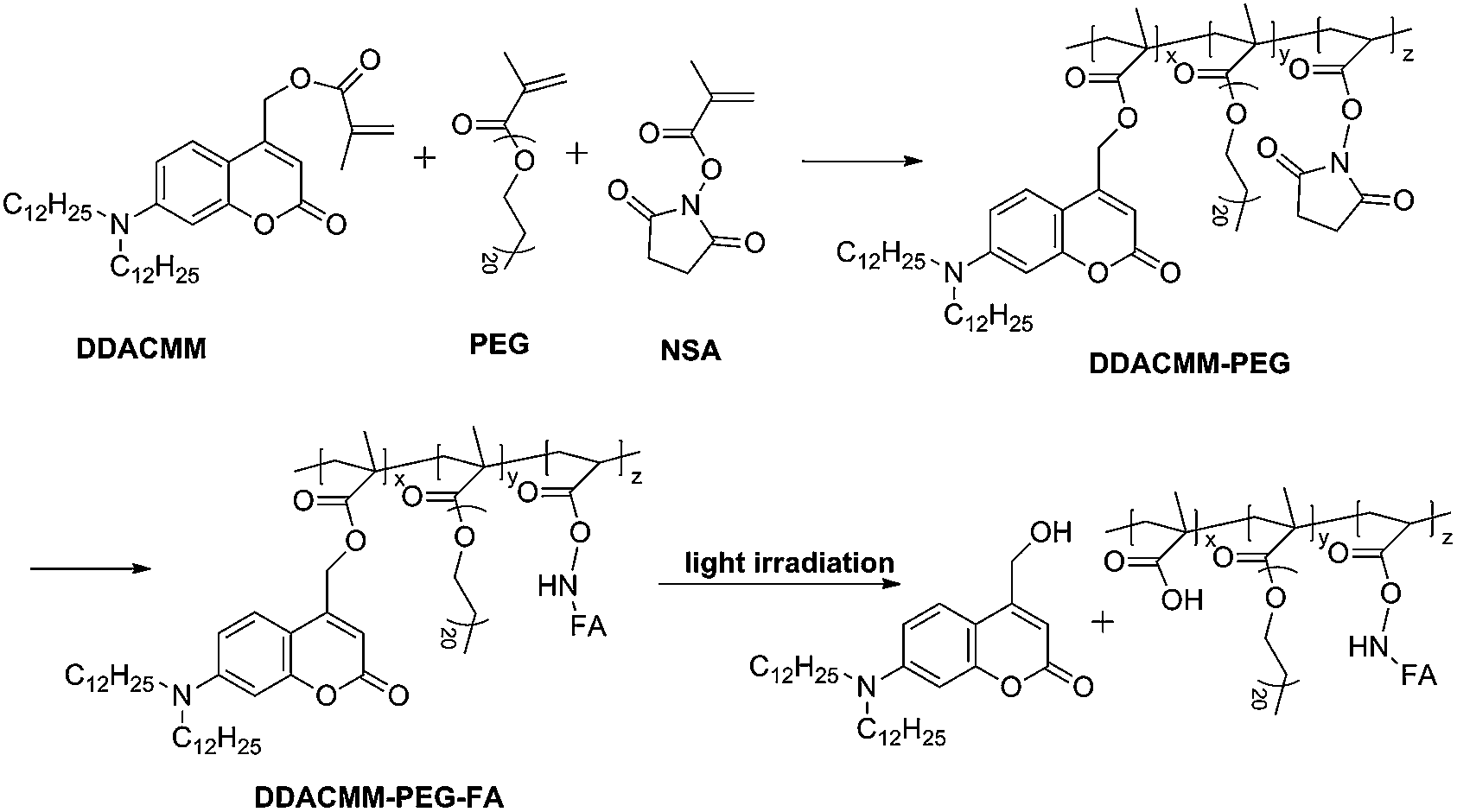 | ||
| Scheme 2 Schematic synthesis process of DDACMM-PEG-FA. | ||
To obtain the HPFe3O4, we first synthesized the amorphous shell–core Fe3O4/Fe (Fig. 1a), and then oxidized the Fe core. The resultant HFe3O4 have a Fe3O4 shell with a thickness of about 4–5 nm and a hollow interior about 10 nm in diameter (Fig. 1b). The small Fe3O4 grains in the shell tend to grow into larger crystallines after treated at the high temperature, leading to the wider gap between two Fe3O4 crystal domains and the formation of pores in the shell (Fig. 1d). The monodisperse HPFe3O4 with an average diameter of 18 nm (Fig. 1c) were prepared successfully, and the XRD powder pattern of HPFe3O4 reveals the face-centered cubic magnetite (Fe3O4) structure (Fig. S3†). The N2 adsorption–desorption isotherms of HPFe3O4 showed in Fig. S4† possesses a typical type IV isotherm with a leap start point at a relative pressure of p/p0 = 0.4. Meanwhile, a distinctively larger hysteresis loop appears in its isotherm, which represents ink-bottle-type pores where larger cavities are connected by narrow windows.
 | ||
| Fig. 1 TEM images of Fe3O4/Fe (a), HFe3O4 (b), HPFe3O4 (c) dispersed in hexane and HPFe3O4@DDACMM-PEG-FA in water. | ||
Fabrication of HPFe3O4@DDACMM-PEG-FA via self-assembly
Because the HPFe3O4 was stabilized by oleic acid, the HPFe3O4 could be easily coated with hydrophobic portions of amphipathic copolymers through the strength of van der Waals forces via a simple self-assembly strategy. The detailed morphological and structure features of the prepared nanoparticles were determined by TEM. In the TEM image of HPFe3O4@DDACMM-PEG-FA (Fig. 1d), we can see the polymer has coated HPFe3O4 successfully.Evaluation of drug loading and release
First, in order to evaluate what HPFe3O4 effects on the polymer disruption, we had detected the fluorescence of polymer (DDACMM-PEG-FA), polymer-coated HPFe3O4, polymer under NIR light exposure and polymer-coated HPFe3O4 under NIR light exposure. As the curves shown in Fig. 2, the HPFe3O4 core has no effects on the fluorescence or degradation of the polymer we prepared.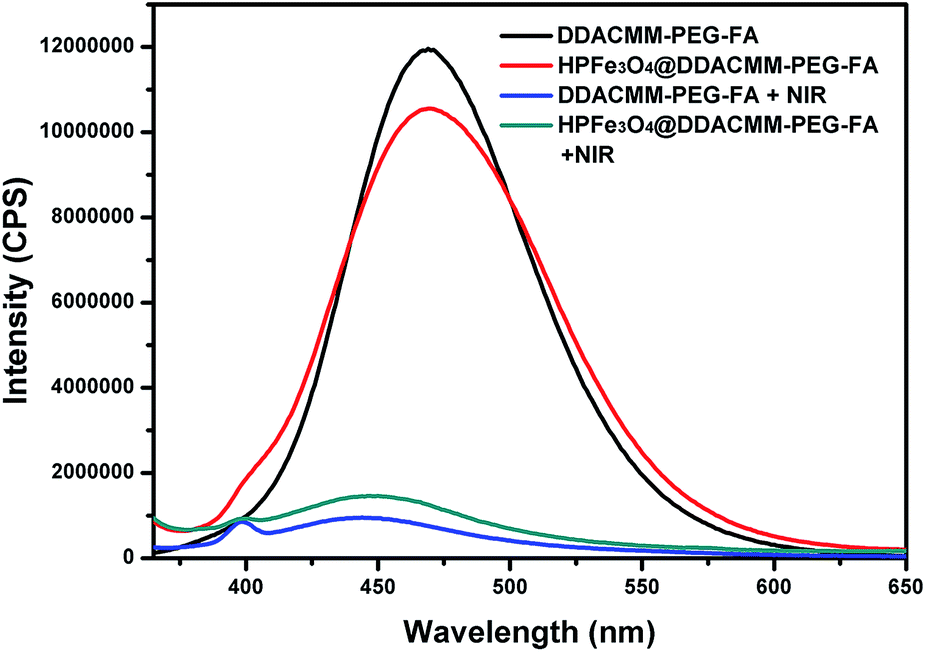 | ||
| Fig. 2 The fluorescence of polymer (DDACMM-PEG-FA), polymer-coated HPFe3O4, polymer under NIR light exposure and polymer-coated HPFe3O4 under NIR light exposure. | ||
Here, we used doxorubicin (DOX) as a model anticancer agent to evaluate the drug loading and release efficiency of the HPFe3O4@DDACMM-PEG-FA. The drug loading content was determined by measuring drug concentration before and after being loaded by HPFe3O4 using a fluorescence spectrophotometer at λex = 475 nm and λem = 592 nm. And a standard curve had been drawn under identical conditions in advance to measure DOX concentration. The theoretical drug loading contents were set at 5%, 10% and 50%. DOX loading efficiency was accordingly measured as 79.40%, 72.30% and 65.86% (Table S2†).
To evaluate the drug release process, we analyzed the hydrolysis profiles of DDACMM-PEG-FA under UV or NIR light exposure (Fig. 3a), which were determined by UV-Vis spectrum at λ = 385 nm. We found that in the condition of the femtosecond pulse NIR laser (800 nm, 1.3–1.5 W), the polymer can be easily hydrolyzed as same as under UV (365 nm, 125 W) exposure, and only a little change was observed under continuous-wave diode NIR laser (808 nm, 4 W). Since the better biocompatibility of the NIR light and well degradation efficiency, a femtosecond pulse NIR laser was required for subsequent experiment. The in vitro cumulative DOX release profiles of the nanocarrier were investigated at 37 °C and under NIR light exposure as shown in Fig. 3b. It was seen that after NIR irradiation, the absorption of DOX increased continuously over time, indicating significant release from the nanocarriers. Meanwhile, there was much more DOX (about 20% increase in 24 h) released from nanocarriers in the solution of pH 5.0 than that of pH 7.0, which confirmed our thought of the pore etching under acid condition. However, without NIR irradiation, even in the condition of pH = 5.0, little absorption of DOX could be detected.
 | ||
| Fig. 3 The degradation degree of DDACMM-PEG-FA under different light exposure (a) and the cumulative release of DOX in vitro from HFe3O4@DDACMM-PEG-FA under femtosecond pulse NIR laser exposure or in dark (b). | ||
Evaluation of cytotoxicity and uptake
We used the MTT assay to assess the cytotoxicity of the HPFe3O4@DDACMM-PEG-FA at various conditions. We incubated KB cells in the culture medium with NIR excitation (800 nm, 1 kHz, 1.3–1.5 W) for 30 min, DOX-loaded HPFe3O4@DDACMM-PEG-FA (DOX-loaded nanocarrier), DOX-loaded nanocarrier under NIR excitation and with nanocarrier under NIR excitation as the controls and then the cells were further incubated for 24 h in the dark. In this assay, the cells under irradiation of NIR light for 30 min showed little toxicity of NIR. The cell viability of cells cultivated by DOX-loaded nanocarrier without light irradiation and nanocarrier with light irradiation both maintained in a high level, which confirmed the safety and biocompatibility of the nanoparticle itself or the residue after loaded drug release. In contrast, the DOX-loaded nanoparticles upon irradiation at 800 nm showed a significantly enhanced cytotoxicity to the cells, which could be explained by the controlled drug release and significant accumulation of DOX inside the cells (Fig. 4a). | ||
| Fig. 4 Inhibition of KB cell growth with excitation by a femtosecond pulse NIR laser (30 min), and DOX-loaded nanocarrier with or without NIR light exposure and nanocarrier with NIR light exposure after 24 h of incubation (100 μL of 1 mg mL−1 the materials solution was added, the NIR exposure at every time was 30 min) (a), and a concentration dependence test for cancer cell ablation (100 μL, 200 μL, 300 μL, 400 μL of 1 mg mL−1 the materials solution was added) under NIR light exposure for 30 min (b). | ||
To evaluate the efficacy of the nanomaterials, DOX-loaded nanocarriers with different quantity was added to incubate KB cells. Then the cells were excited under NIR light exposure. From Fig. 4b, we could find out that the inhibition efficacy to the cancer cells has a linear relationship with the dosage of nanocarriers.
Since the polymer contains coumarin fluorescent moieties, the fluorescence intensity could be used to indicate the intracellular activity of the nanoparticles in FR(+) (KB cells) cancer cell line. Here, in vitro experiments based on this cell were used to prove the targeting properties of the nanocarriers. The KB cell uptake and intracellular distribution of the nanocarriers with (HPFe3O4@DDACMM-PEG-FA) (Fig. 5a and b) and without (HPFe3O4@DDACMM-PEG) FA groups (Fig. 5c and d) for 0.5 and 1 h were studied by fluorescence microscopy.
 | ||
| Fig. 5 CLSM of HPFe3O4@DDACMM-PEG-FA (with FA targeting groups) in KB cells cultivated for 30 min (a) and 1 h (b), HPFe3O4-DDACMM-PEG (without FA targeting groups) in KB cultivated for 30 min (c) and 60 min (d). Mean fluorescence intensity on KB cells incubated HPFe3O4@DDACMM-PEG-FA (with FA targeting groups) and HPFe3O4-DDACMM-PEG (without FA targeting groups) at different incubation times (e). | ||
From the photos using confocal laser scanning microscope (CLSM), a brighter image in fluorescence intensity, not merely after 0.5 h but also 1 h, can be seen in KB cells incubated with FA-conjugated nanocarriers than the other. This result demonstrated that the targeting moiety offered by folic acid is efficient at enhancing the tumor cell targeting in vitro because the FA-conjugated nanocarrier was taken up by KB cells through a folate receptor-mediated endocytosis.
Magnetic properties
In view of the low cytotoxicity and high colloidal stability, the nanocarrier could be a safe MRI contrast agent in vivo. Fig. 6a shows a T2-weighted image changed remarkably in signal intensity with increasing Fe concentration of the nanocarriers, and accordingly, the relaxivity (1/T2) increases linearly (Fig. 6b). The specific relaxivity (r2) was calculated to be 139.1 Fe mM−1 s−1. The picture and curve indicated the HPFe3O4 generated MRI contrasts on T2-weighted spin-echo sequences and HPFe3O4@DDACMM-PEG-FA could be a great T2 contrast agent. In addition, MR imaging was further used to confirm the target ability of HPFe3O4@DDACMM-PEG-FA. The T2-weighted MR imaging (Fig. S6†) of KB cells incubated with HPFe3O4@DDACMM-PEG-FA showed an obvious negative contrast enhancement in comparison to the HPFe3O4@DDACMM-PEG, which revealed effective MRI efficacy and target ability of the nanocarriers inside the KB cells. | ||
| Fig. 6 T2-weighted MR images (a) and relaxation rates (1/T2) of the aqueous dispersion of HPFe3O4@DDACMM-PEG-FA at various concentrations (b). | ||
Conclusions
In summary, we have fabricated a NIR light and pH dual responsive nanocarrier for anti-cancer delivery via a simple self-assembly strategy. In this strategy, we used hollow porous Fe3O4 (HPFe3O4) as “core” whose pore size could be enlarged in weak acid environment. A FA-conjugated NIR light responsive polymer was then coated on the HPFe3O4. Thus, we got NIR light- and pH-responsive nanocarriers by self-assembly. NIR light could be used to control the degradation of the polymer outside and the subacid environment in tumour could improve the efficiency of the drug release (about 20% increases in 24 h). The as-prepared nanocarrier could not only act as an efficient drug carrier, but could also be used to track the cancer targeting process due to its magnetism and fluorescence. We are convinced that this smart drug nanocarrier is potentially useful for cancer chemotherapy.Acknowledgements
We gratefully acknowledge the financial support provided by National Natural Science Foundation of China (21336005, 21301125), Natural Science Foundation of Jiangsu Province (BK2012625), Natural Science Foundation of the Jiangsu Higher Education Institutions of China (13KJB430022) and Joint Research Projects of SUN-WIN Joint Research Institute for Nanotechnology.Notes and references
- Z. T. Luo, K. Y. Zheng and J. P. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- D. G. Yu, G. R. Williams, X. Wang, X. K. Liu, H. L. Li and S. W. Annie Bligh, RSC Adv., 2013, 3, 4652–4658 RSC.
- J. P. Xie, Q. B. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2(12), 2473–2480 CrossRef CAS PubMed.
- M. G. Carstens, C. F. van Nostrum, R. Verrijk, L. G. J. De Leede, D. J. A. Crommelin and W. E. Hennink, J. Pharm. Sci., 2008, 97, 506 CrossRef CAS PubMed.
- N. Kasyapi and A. K. Bhowmick, RSC Adv., 2014, 4, 27439–27451 RSC.
- D. Y. Chen, X. W. Xia, H. W. Gu, Q. F. Xu, J. F. Ge, Y. G. Li, N. J. Li and J. M. Lu, J. Mater. Chem., 2011, 21, 12682 RSC.
- Y. Wang, Y. Xiao, H. Y. Zhou, W. Chen and R. K. Tang, RSC Adv., 2013, 3, 23133–23138 RSC.
- W. Wu, M. Chen, J. T. Wang, Q. J. Zhang, S. Li, Z. F. Lin and J. S. Li, RSC Adv., 2014, 4, 30780–30783 RSC.
- J. L. Li, X. Q. An, Z. F. Pan and L. M. Sun, RSC Adv., 2014, 4, 9476–9479 RSC.
- S. Sortino, J. Mater. Chem., 2012, 22, 301–318 RSC.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- X. Mei, S. Yang, D. Y. Chen, N. J. Li, H. Li, Q. F. Xu, J. F. Ge and J. M. Lu, Chem. Commun., 2012, 48, 10010–10012 RSC.
- F. L. Callari, S. Petralia, S. Conoci and S. Sortino, New J. Chem., 2008, 32, 1899–1903 RSC.
- A. Fraix, N. Kandoth, I. Manet, V. Cardile, A. C. E. Graziano, R. Gref and S. Sortino, Chem. Commun., 2013, 49, 4459–4461 RSC.
- G. Y. Liu, C. J. Chen, D. D. Li, S. S. Wang and J. Ji, J. Mater. Chem., 2012, 22, 16865–16871 RSC.
- T. Ribeiro, S. Raja, A. S. Rodrigues, F. Fernandes, J. P. S. Farinha and C. Baleizão, RSC Adv., 2013, 3, 9171–9174 RSC.
- T. Fujigaya, T. Morimoto and N. Nakashima, Soft Matter, 2011, 7, 2647–2652 RSC.
- Q. N. Lin, C. Y. Bao, S. Y. Cheng, Y. L. Yang, W. Ji and L. Y. Zhu, J. Am. Chem. Soc., 2012, 134, 5052–5055 CrossRef CAS PubMed.
- J. Babin, M. Pelletier, M. Lepage, J. F. Allard, D. Morris and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3329–3332 CrossRef CAS PubMed.
- D. Collado, Y. Vida, F. Najera and E. Perez-Inestrosa, RSC Adv., 2014, 4, 2306–2309 RSC.
- W. D. Ji, N. J. Li, D. Y. Chen, X. X. Qi, W. W. Sha, Y. Jiao, Q. F. Xu and J. M. Lu, J. Mater. Chem. B, 2013, 1, 5942–5949 RSC.
- L. Wang, M. Wang, P. D. Topham and Y. Huang, RSC Adv., 2012, 2, 2433–2438 RSC.
- C. J. Xu and S. H. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed.
- K. G. Neoh and E. T. Kang, Soft Matter, 2012, 8, 2057–2069 RSC.
- F. Yang, Y. Li, Z. Chen, Y. Zhang, J. Wu and N. Gu, Biomaterials, 2009, 30, 3882–3890 CrossRef CAS PubMed.
- M. Vaccaro, G. Mangiapia, A. Radulescu, K. Schillén, G. D'Errico, G. Morelli and L. Paduano, Soft Matter, 2009, 5, 2504–2512 CAS.
- Q. Li, L. F. Zhang, L. J. Bai, Z. B. Zhang, J. Zhu, N. C. Zhou, Z. P. Cheng and X. L. Zhu, Soft Matter, 2011, 7, 6958–6966 RSC.
- M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldöfner, R. Scholz, S. Deger, P. Wust, S. A. Loening and A. Jordan, Int. J. Hyperthermia, 2005, 21, 637–647 CrossRef CAS.
- M. Johannsen, U. Gneveckow, K. Taymoorian, B. Thiesen, N. Waldöfner, R. Scholz, K. Jung, A. Jordan, P. Wust and S. A. Loening, Int. J. Hyperthermia, 2007, 23, 315–323 CrossRef CAS PubMed.
- H. X. Wu, S. J. Zhng, J. M. Zhang, G. Liu, J. L. Shi, L. X. Zhang, X. Z. Cui, M. L. Ruan, Q. J. He and W. B. Bu, Adv. Funct. Mater., 2011, 21, 1850–1862 CrossRef CAS.
- S. Peng, C. Wang, J. Xie and S. H. Sun, J. Am. Chem. Soc., 2006, 128, 10676–10677 CrossRef CAS PubMed.
- S. Peng and S. H. Sun, Angew. Chem., Int. Ed., 2007, 46, 4155–4158 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. J. Xu and S. H. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- D. M. Mizrahi, M. Omer-Mizrahi, J. Goldshtein, N. Askinadze and S. Margel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5468–5478 CrossRef CAS.
- Y. Pan, J. H. Gao, B. Zhang, X. X. Zhang and B. Xu, Langmuir, 2010, 26, 4184–4187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07573k |
| This journal is © The Royal Society of Chemistry 2014 |