DOI:
10.1039/C4RA07554D
(Paper)
RSC Adv., 2014,
4, 48177-48190
Mesoporous poly-melamine-formaldehyde stabilized palladium nanoparticle (Pd@mPMF) catalyzed mono and double carbonylation of aryl halides with amines†
Received
24th July 2014
, Accepted 22nd September 2014
First published on 23rd September 2014
Abstract
A new mesoporous poly-melamine-formaldehyde material supported Pd nano catalyst (mPMF–Pd0) has been synthesized and characterized by thermogravimetric analysis (TGA), powder X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray spectrometry (EDS), high-resolution transmission electron microscopy (HRTEM), UV-vis diffuse reflection spectroscopy (DRS), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS) and a N2 adsorption study. The mPMF–Pd0 material showed very good catalytic activity in the field of mono and double amino carbonylation of aryl bromides/iodides. Moreover, the catalyst is easily recoverable and can be reused six times without appreciable loss of catalytic activity in the above reactions. So, the highly dispersed and strongly bound palladium(0) sites in the mPMF–Pd0 could be responsible for the observed high activities. Due to strong binding with the functional groups of the polymer, no evidence of leached Pd from the catalyst during the course of reaction occurred, suggesting true heterogeneity in the catalytic process.
1. Introduction
Metal nanoparticles have gained substantial attention in recent years in both chemistry and biology.1,2 The smaller size of the metal nanoparticles leads to a high surface/volume ratio and consequently, a large number of potential active sites would be available to the substrates, thereby enhancing their catalytic activity. However, metal nanoparticles, due to their high surface energy, are not stable as such. For instance, palladium nanoparticles aggregate into Pd black, which possesses lower catalytic activity.3 Hence immobilization of metal nanoparticles into a suitable rigid matrix has come into practice. In particular, Pd nanoparticles are stabilized by dispersing them in porous materials, ionic liquids, surfactants, dendrimers, functionalized polymers, biopolymers, various ligands, silica in supercritical CO2, etc.4,5 The stabilizing medium serves as a supporting material for protecting the nanoparticles from aggregation, provides the desired chemical interface between the nanoparticles and substrate, prevents the metal from being leached out of the matrix during the reaction and thus facilitates the overall reaction.
Various supports such as silica, carbon, and zirconia have been used for the immobilization of palladium nano particles.6–8 These supports, though advantageous in some cases, retain some of the common drawbacks that pertain to the traditional and cumbersome isolation procedure after completion of reactions and most importantly are deficient in the reusability aspect. However, ordinary mesoporous materials are poor in functional groups and the impregnation of metal nanoparticles often results in polydisperse nanoparticles on the supportive materials. Even so, there is an ample scope in the design of improved supported catalysts from the point of view of benign and sustainable protocols.
Among the different type of mesoporous materials, the organic mesoporous polymers are a class of advanced materials, which possess the textural porosities of mesoporous materials (well-ordered pores, large surface area and tunable structures) and simultaneously the advantages of organic polymers (high hydrophobicity, containing aromatic sections and higher stability in acid or base media in comparison to silica based materials).9,10 Recently, we have reported several mesoporous organic polymer supported metal catalysts.11
In this context, mesoporous organic polymers carrying the reactive functional groups at the surface of the mesopores12–16 can provide an ideal tethering agent for the active metals to bind at its surface strongly. Due to extensive cross-linking, polymeric materials are highly desirable material for long term stabilization of the entrapped metal centres, which reduces the possibility of leaching of the metal under reaction conditions.17
On the other hand, the palladium-catalyzed carbonylation of aryl halides in the presence of carbon monoxide is an important methodology for the preparation of carbonyl containing derivatives.18 The amides and α-ketoamides are important targets in organic synthesis because of their ability to serve as building blocks in a wide variety of functional group transformations.19 They have also been found as a substructure in numerous bioactive natural products and pharmaceutically interesting compounds.20 Therefore, the development of methods that can construct these two classes of nitrogen containing compounds in an efficient and regioselective manner from readily accessible substrates continues to be a hot research topic.
Several strategies have been reported in the literature to achieve their synthesis21 among which the transition metal catalysis approaches have proven to be the most effective for providing access to the desired compounds in good yields and high selectivities. Although, few examples have been reported on mono and double carbonylation of aryl halides with amines for the synthesis of amides and α-ketoamides respectively.22
Most of the works have been realised in the presence of homogeneous Pd catalysts making the separation/purification and recovery of the expensive metal catalysts very difficult, or even impossible. These environmental and economic concerns associated with the contamination issues of biologically active compounds are particularly important when dealing with large scale-synthesis and industrial processes. Although few examples have been reported for mono carbonylation, reports on double carbonylation of aryl halides with amine catalysed by heterogeneous palladium nano materials are still rare.23 Less expensive aryl bromides are more attractive substrates for the amino carbonylation but more expensive aryl iodides are generally the substrate of choice in amino carbonylation catalyzed by homo or heterogeneous Pd catalyst and most of these reported methods require harsher conditions.22 It is thus desirable to develop an efficient, one-pot, cost effective, phosphine free and green methodology to avoid all these difficulties.
Herein, we report a novel mesoporous poly-melamine-formaldehyde (mPMF) material24 supported Pd nano catalyst that exhibits superb activity in catalyzing the aryl bromides for the synthesis of aryl amides and this novel Pd catalyst is also highly effective for the synthesis of α-ketoamides from aryl iodides under phosphine free condition. The high activity of mPMF–Pd0 is attributed to its condensed network structure which contains large number of nitrogen atoms. Significant amounts of nitrogen have been incorporated into comparable inorganic framework materials25 and this is found to be beneficial for the stabilization of Pd nano.
2. Experimental section
2.1. Chemicals
Melamine and paraformaldehyde were purchased from Sigma-Aldrich. Pd(OAc)2 was purchased from Universal Chemicals, India. Sodium borohydride (NaBH4) was obtained from Spectrochem, India and used as received. All other reagents and substrates were purchased from Merck, India and were used without further purification. Solvents were dried and distilled through standard procedure.
2.2. Physical measurements
The FT-IR spectra of the samples were recorded from 400 to 4000 cm−1 on a Perkins Elmer FT-IR 783 spectrophotometer using KBr pellets. UV-vis spectra were taken using a Shimadzu UV-2401PC doubled beam spectrophotometer having an integrating sphere attachment for solid samples. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo TGA/DTA 851e. Surface morphology of the samples was measured using a scanning electron microscope (SEM) (ZEISS EVO40, England) equipped with EDX facility. Specific surface area of the sample was measured by adsorption of nitrogen gas at 77 K and applying the Brunauer–Emmett–Teller (BET) calculation. Prior to adsorption, the samples were degassed at 250 °C for 3 h. Powder X-ray diffraction (XRD) patterns of different samples were analyzed with a Bruker D8 Advance X-ray diffractometer using Ni-filtered Cu Kα (λ = 0.15406 nm) radiation. Transmission electron microscopy (TEM) images of the mesoporous polymer were obtained using a JEOL JEM 2010 transmission electron microscope operating at 200 kV. Elemental analysis was carried out in a 2400 Series-II CHN Analyzer, Perkin–Elmer, USA. NMR spectra were recorded on a Varian Mercury plus NMR spectrometer in pure deuterated solvents.
2.3. Synthesis of mesoporous poly-melamine-formaldehyde polymer (mPMF)
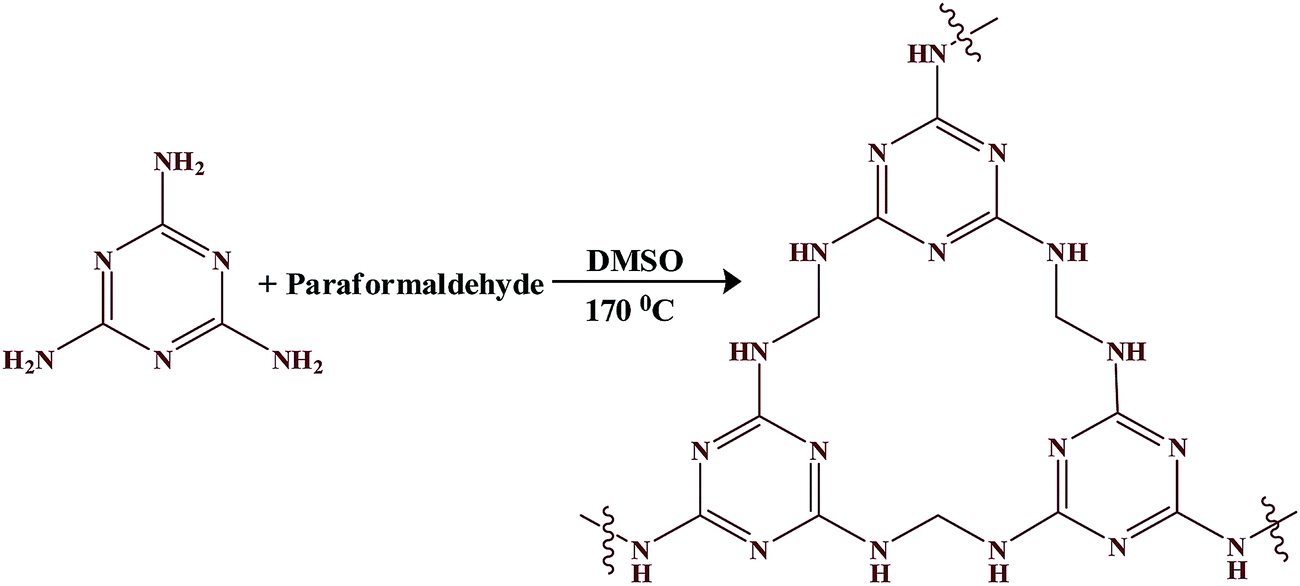
Melamine (0.378 g, 3 mmol) and paraformaldehyde (1.8 eq., 0.162 g, 5.4 mmol) were mixed with 3.36 mL (overall concentration of 2.5 M) of dimethyl sulfoxide (DMSO) in a 15 mL Teflon container secured in a steel reactor. The reaction mixture was heated to 120 °C in an oven for 1 h. The reactor was then carefully removed from the oven for stirring on a magnetic stirrer plate to obtain a homogeneous solution. It was then heated in the oven to 170 °C for 72 h. The reaction was allowed to cool to room temperature, and the obtained solid was crushed, filtered, and washed with DMSO, acetone, tetrahydrofuran (THF) and CH2Cl2. The resulting white solid was dried under vacuum at 80 °C for 24 h (Scheme 1).
 |
| | Scheme 1 Schematic diagram showing the formation of mesoporous polymer. | |
2.4. Synthesis of colloidal Pd nanoparticles
In a typical synthesis, 150 mg of Pd(OAc)2 was added to 10 mL of distilled water containing 0.5 mmol Tris-buffer and was stirred for 2 min. Then, aqueous solution of NaBH4 (37 mg, 1 mmol) was added drop-wise under stirring. The stirring was continued for another 10 min, and the resulting nanocolloid was stored at 4 °C.
2.5. Synthesis of mPMF–Pd0 nanocatalyst
A total of 600 mg of mPMF was dispersed in 10 mL of TRIS-stabilized Pd-NPs and stirred for 1 h at room temperature. The colour of the colloidal nanoparticles gradually disappeared while stirring. The supernatant solution was colourless after 1 h of stirring at room temperature, whereas the colour of mPMF changed to black, indicating the loading of Pd-NPs onto the surface of mPMF. After centrifugation, black coloured Pd-NP containing mesoporous polymer mPMF–Pd0 was obtained. This mesoporous material was washed further with copious amounts of water and dried at room temperature. Pd(0) loading to the mPMF was determined to be 10.0% by AAS (Scheme 2).
 |
| | Scheme 2 Schematic diagram showing the formation of mesoporous mPMF–Pd0 nanocatalyst. | |
2.6. General procedure of mono-carbonylation of aryl bromides with morpholine
A 50 mL high-pressure reactor was charged with toluene (3.0 mL), K3PO4 (2.5 equiv.), aryl bromides (1 mmol), morpholine (1.5 mmol), mPMF–Pd0 (25 mg) and CO (3 atm) before heating at 80 °C for 10 h. After cooling the reactor, the CO was vented from the reactor and the desired compound was purified by chromatography on silica gel using a pentane/CH2Cl2 eluent system. All the prepared compounds were confirmed by 1H NMR and 13C spectrum.
2.7. General procedure of double-carbonylation of aryl iodides with amines
A 50 mL high-pressure reactor was charged with DMF (3.0 mL), K2CO3 (2.5 equiv.), aryl iodides (1 mmol), morpholine (1.5 mmol), mPMF–Pd0 (25 mg) and CO (20 atm) before heating at 90 °C for 12 h. After cooling the reactor, the CO was vented from the reactor and the desired compound was purified by chromatography on silica gel using a pentane/CH2Cl2 eluent system. All the prepared compounds were confirmed by 1H and 13C NMR spectrum.
3. Results and discussion
3.1. Characterization of Pd–mPMF material
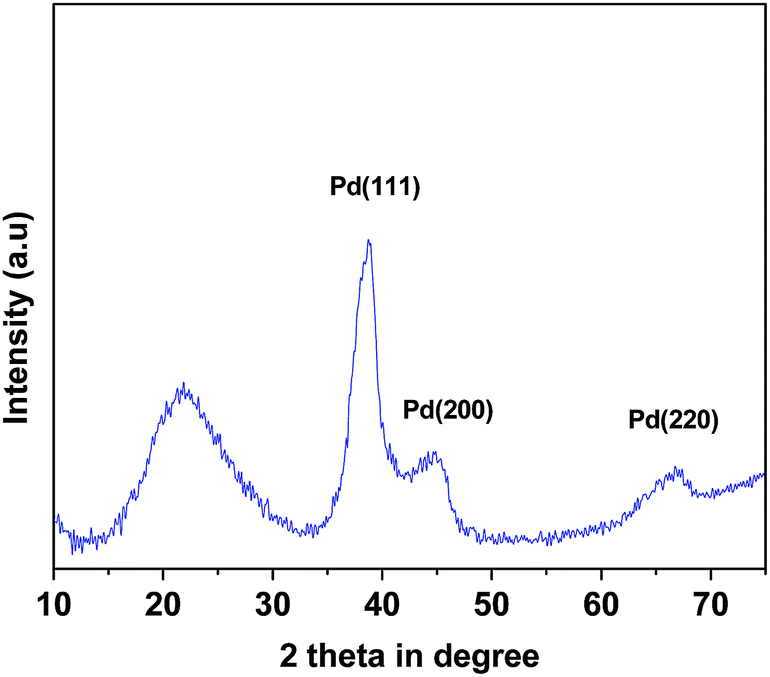
X-ray diffraction. In Fig. 1 powder X-ray diffraction pattern of mPMF–Pd0 is shown. The mesoporous polymer, mPMF shows one broad diffraction peak cantered at 2θ = 21.70, which correspond to characteristic peak of the mPMF. In the XRD pattern of the mPMF–Pd0 shows the additional diffraction peaks at 2θ = 38.80, 44.85 and 67.00 degree are attributed to Pd nano.26
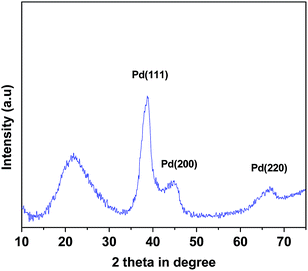 |
| | Fig. 1 Powder XRD pattern of mPMF–Pd0 material. | |
HR-TEM analysis. Fig. 2 demonstrates HR-TEM images of Pd-NPs supported on mesoporous poly-melamine-formaldehyde. HR-TEM images (Fig. 2a and b) suggested a foam-like interconnected mesoporous network structure contained high electron density dark spots of size 1.5 to 2 nm (ref. 26) throughout the specimen, which are spherical in nature (Fig. 2c). These spherical particles are assigned to Pd-nanoparticles in mPMF–Pd0. TEM-EDS from the obtained nanomaterials (Fig. 3) provided the presence of the expected elements in the structure of the catalyst, namely palladium, nitrogen and carbon.
 |
| | Fig. 2 HR-TEM images of mPMF–Pd0 material. | |
 |
| | Fig. 3 TEM-EDS image of mPMF–Pd0 material. | |
SEM analysis. The scanning electron micrograph of the mPMF–Pd0 is shown in Fig. 4. The FE-SEM image (Fig. 4A) indicates uniform submicronsized spherical morphology of the polymer mPMF–Pd0 material. Spherical particles get aggregated among themselves to form large assembly of particles and these are inter connected with each others (Fig. 4B).
 |
| | Fig. 4 SEM images of mPMF–Pd0 material. | |
Raman spectroscopy. In addition to the optical spectroscopy, Raman spectroscopy also provides useful information regarding the interaction of PdNPs and mesoporous mPMF. Raman spectra of the mPMF–Pd0 shows two well documented D and G band at 1362 and 1549 cm−1 respectively (Fig. 5). The D band at 1362 cm−1 is attributed due to the defect or disorder in carbon atom and G band at 1549 cm−1 is attributed due to sp2 in plane vibration of carbon atom. Raman spectroscopy of mesoporous mPMF–Pd0 showed different intensity peaks from each component. The peak at 482 cm−1 corresponded to Pd nano and the peak at 1057 cm−1 corresponded to the T band.27
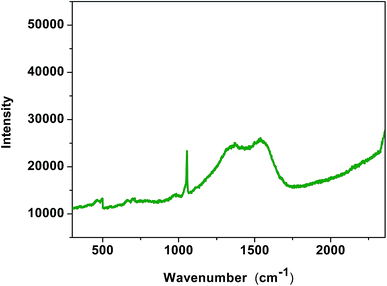 |
| | Fig. 5 Raman spectroscopy of mPMF–Pd0. | |
XPS analysis. X-ray photoelectron spectroscopy provides the valuable information regarding the oxidation state of Pd. The XPS spectrum of mPMF–Pd0 was shown in the Fig. 6. Two main peaks related to Pd 3d5/2 and Pd 3d3/2 is centered at the XPS binding energies of 337.07 eV and 342.57 eV respectively in the XPS spectrum of mPMF–Pd0. This XPS data clearly reveals that the Pd(0) species have been generated during the course of reduction, which are embedded uniformly throughout the mesoporous material. These binding energy values agree well with the values for palladium(0) state accounted into the literature. No peak corresponding to Pd2+ in the XPS spectrum was found, which discloses the fact that the palladium(II) has been completely reduced into metallic palladium nanoparticles during the reduction technique.28
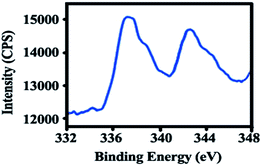 |
| | Fig. 6 XPS of mPMF–Pd0. | |
3.2. Catalytic activities
Since mesoporous supported palladium nano exhibit high catalytic activity in a wide range of industrially important processes and have been extensively studied for c–c coupling, thus we decided to investigate the catalytic activity of the mPMF–Pd0 in the field of carbonylation reactions. Carbonylation of aryl halides with secondary amines usually gives rise to a mixture of mono and dicarbonylated products. Chemoselectivity of the reaction is greatly dependent on the reaction conditions: solvent, base, nature of the nucleophile, the catalyst and most importantly the pressure of CO (Scheme 3).
 |
| | Scheme 3 Mono and double carbonylation of aryl halide with morpholine. | |
Mono carbonylation of aryl bromides for the synthesis of aryl amides. To test the catalytic activity of the present catalyst, the carbonylation of aryl bromides were performed (Scheme 4). In this reaction aryl bromide and secondary amine were converted to aryl amide under the reaction conditions. Quantitative conversion of aryl bromide to aryl amide can be achieved by using the mPMF–Pd0 catalyst. The carbonylation reaction of aryl bromides with morpholine was run in a 50 mL autoclave at 80 °C under 3 atm carbon monoxide atmospheres for 10 h reaction time. In our initial studies, the bromobenzene was chosen for the optimization of the mono carbonylation reaction.
 |
| | Scheme 4 Mono carbonylation of bromobenzene with morpholine. | |
The performance of palladium-catalyzed carbonylation reaction is known to be governed by a number of reaction parameters. To find the appropriate reaction conditions various bases and solvents were screened (Table 1). In order to identify effective conditions for promoting the monocabonylation, the reaction was carried out under various catalyst amount and reaction time (Table 2).
Table 1 Effect of base and solvent on mono carbonylationa
| Entry |
Base |
Solvent |
Conversion (%) |
| Reaction conditions: bromobenzene (1.0 mmol), morpholine (1.5 mmol), base (2.5 mmol), CO (3 atm), mPMF–Pd0 (25 mg), toluene (3.0 mL), 80 °C, 10 h. |
| 1 |
K3PO4 |
Toluene |
92 |
| 2 |
K3PO4 |
Dioxane |
37 |
| 3 |
K3PO4 |
DMF |
45 |
| 4 |
K2CO3 |
Toluene |
86 |
| 5 |
K2CO3 |
Dioxane |
28 |
| 6 |
K2CO3 |
DMF |
31 |
| 7 |
Et3N |
Toluene |
21 |
| 8 |
Et3N |
Dioxane |
NR |
| 9 |
Et3N |
DMF |
NR |
Table 2 Effect of time and catalyst amount on mono carbonylationa
| Entry |
Time (h) |
Catalyst (mg) |
Yield (%) |
| Reaction conditions: bromobenzene (1.0 mmol), morpholine (1.5 mmol), K3PO4 (2.5 mmol), CO (3 atm), mPMF–Pd0 (25 mg), toluene (3.0 mL) at 80 °C for 10 h. |
| 1 |
5 |
25 |
34 |
| 2 |
8 |
25 |
68 |
| 3 |
10 |
25 |
92 |
| 4 |
12 |
25 |
92 |
| 5 |
10 |
10 |
29 |
| 6 |
10 |
15 |
45 |
| 7 |
10 |
20 |
67 |
| 8 |
10 |
30 |
92 |
The influence of base on the catalytic performance of this system was investigated by employing various bases. It was observed that inorganic bases are more effective than organic base like Et3N. Within a short time the reaction proceeded in high yield in presence of K3PO4 or K2CO3. We found that using K3PO4 as base in toluene at 80 °C gave the 92% conversion. Et3N was not as effective as K3PO4, only affords to low yield of coupling product. To verify the solvent effect, a series of solvents were investigated for the mono carbonylation of bromobenzene with morpholine as the model reaction. The reaction conducted in nonpolar solvent medium like toluene, was found to be the most effective. The use of DMF or dioxane as solvents led to slower reactions. Consequently, toluene was chosen as the medium of choice for this mono carbonylation.
This coupling reaction was found to be highly sensitive to the catalyst amount. Profound increase in the yield of the desired product was observed when the catalyst amount was increased from 10 to 25 mg; maximum yield was obtained at 25 mg mPMF–Pd0. Also the reaction was carried out for different time ranging from 5 h to 12 h and it was found that at 10 h the conversion was 92% at given conditions.
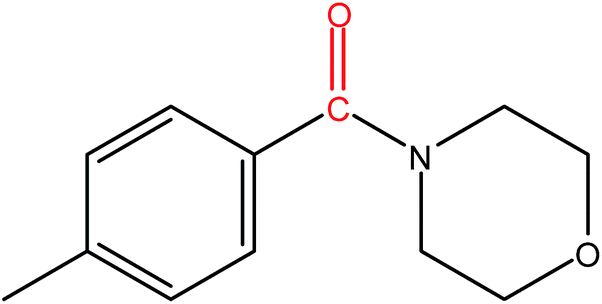
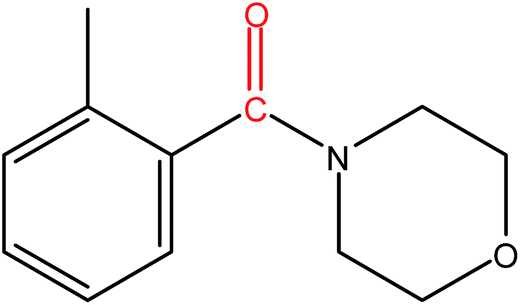
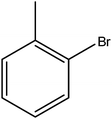
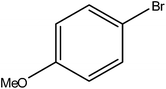
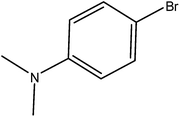
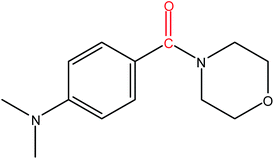
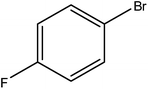
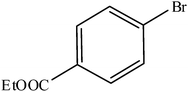
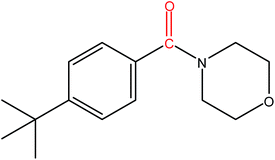
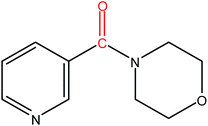
Hence, the finalized reaction conditions were the following: base, K3PO4; temperature, 80 °C; solvent, toluene; time, 10 h; and 3 atm of CO pressure. These reaction parameters were then successfully applied for mono carbonylation of a variety of aryl bromides with morpholine in toluene. The experimental results are summarized in Table 3. With a defined protocol in hand, the substrate scope with various aryl bromides was explored. As shown in Table 3, the protocol well tolerated diverse electronic and steric substituents of the aryl bromides, as well as heterocyclic substrates, all resulting in good to excellent yields. The relative position of substituents slightly impacted the coupling efficiency: p-bromotoluene resulted in a higher yield than its m or o-analogues (Table 3, entries 2–4). The substitution of electron withdrawing and electron donating groups on the phenyl ring of aryl bromides did not have any appreciable influence on the outcome of the reaction. Aryl bromides bearing either electron-donating or electron-withdrawing substituents, afforded the corresponding cabonylative products in good to excellent yields. To our surprise, we found that Strong electron-donating groups such as –OMe and –NMe2 did not influence the activity significantly compared to electron withdrawing groups such as –F and –CF3 (Table 3, entries 8 and 9). To our delight, electron-deficient bromoarenes also provided good to excellent yields (Table 3, entries 8–11). As expected, heterocyclic and bulky bromoarenes all afforded excellent yields (Table 3, entries 15–18).
Table 3 mPMF–Pd0 catalyzed mono carbonylation of aryl bromides with morpholinea
| Entry |
Substrate |
Product |
Yield (%) |
| Reaction conditions: aryl bromide (1.0 mmol), morpholine (1.5 mmol), K3PO4 (2.5 mmol), CO (3 atm), mPMF–Pd0 (25 mg), toluene (3.0 mL) at 80 °C for 10 h. |
| 1 |
 |
 |
92 |
| 2 |
 |
 |
94 |
| 3 |
 |
 |
71 |
| 4 |
 |
 |
83 |
| 5 |
 |
 |
97 |
| 6 |
 |
 |
88 |
| 7 |
 |
 |
70 |
| 8 |
 |
 |
89 |
| 9 |
 |
 |
88 |
| 10 |
 |
 |
85 |
| 11 |
 |
 |
92 |
| 12 |
 |
 |
91 |
| 13 |
 |
 |
80 |
| 14 |
 |
 |
82 |
| 15 |
 |
 |
90 |
| 16 |
 |
 |
92 |
| 17 |
 |
 |
94 |
| 18 |
 |
 |
95 |
Double carbonylation of aryl iodides for the synthesis of α-ketoamides. In continuation of our interest in carbonylation reactions, we have extended the application of our newly prepared mPMF–Pd0 in field of double carbonylation reactions for the synthesis of α-ketoamides from aryl iodides. Our study of double carbonylation reaction began on the basis of the work reported by Marie et al.,29 the reaction was reported in DMF with iodobenzene, morpholine and Et3N as the base at 60 °C under 40 bar of CO. The coupling was carried out in the presence of 1 mol% [Pd] using PdCl2(PPh2)2@SBA-15 as the model catalyst. Under these conditions, high selectivity, up to 74%, for the double carbonylation product was reported. However, the method requires high CO pressure and long reaction time (48 h). Since high-pressure conditions make them expensive for industry and difficult to handle in the laboratory and in industry, this observation makes the present carbonylation catalyst even more attractive. To improve this preliminary result and reaction conditions we have performed the similar reaction catalyzed by our mPMF–Pd0 in DMF at 90 °C and under the 20 atm CO pressures. Delightfully, the desired product was obtained in excellent yield (98%). To optimize the reaction conditions we have screened various solvents and bases and maximum yield was obtained for K2CO3 in DMF. To get maximum selectivity towards double carbonylations we have varied the CO pressure from 1 atm to 25 atm and maximum double carbonylated yield was obtained at 20 atm CO pressure (Fig. 7). The reaction was carried out by varying the CO pressure (1 atm to 25 atm) keeping the other parameters fixed. The reaction mixtures were collected after completion of reaction at different pressure. The products were identified and quantified by GC. All obtained results from each run were plotted in Fig. 7. It has been found that the selectivity of double carbonylation gradually increased at high pressure region.
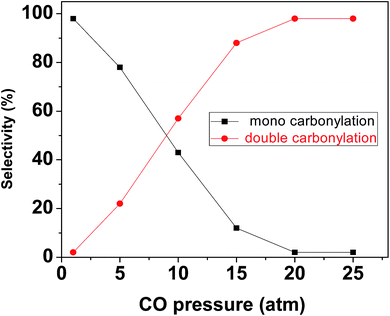 |
| | Fig. 7 Effect of CO pressure on carbonylation of iodobenzene with morpholine. Reaction conditions: iodobenzene (1.0 mmol), morpholine (1.5 mmol), K2CO3 (2.5 mmol), mPMF–Pd0 (25 mg), DMF (5.0 mL) at 90 °C for 12 h. | |
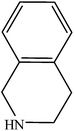
To explore the generality and scope of the double carbonylation reaction, the reaction of various aryl iodides and amines was carried out under optimum conditions (aryl iodide, 1.0 mmol; K2CO3, 2.0 mmol; amines, 5.0 mmol; DMF, 5.0 mL; mPMF–Pd0 25 mg; P(CO) = 20 atm, 90 °C, 12 h) and the results are summarized in Table 4. Various aryl iodides afforded the corresponding α-ketoamides in moderate to good yields. On the other hand, the steric bulkiness of amine has a good conversion and selectivity (Table 4, entries 2–4). Less basic amines such as aniline did not afford any double carbonylated product. It is noteworthy that the α-ketoamides were obtained with good selectivities when using bulky amines such as 1,2,3,4-tetrahydroisoquinoline as nucleophilic reagents (Table 4, entries 3 and 6–10). In an attempt to extend the substrate scope to aryl bromides, we also tested the double carbonylation reaction of bromobenzene with diethylamine. However, no significant conversion was observed.
Table 4 mPMF–Pd0 catalyzed double carbonylation of aryl iodides with aminesa
| Entry |
Aryl iodide |
Amine |
Product |
Yield (%) |
| Reaction conditions: aryl iodides (1.0 mmol), amines (1.5 mmol), base (2.5 mmol), CO (20 atm), mPMF–Pd0 (25 mg), DMF (5.0 mL) at 90 °C for 12 h. |
| 1 |
 |
 |
 |
98 |
| 2 |
 |
 |
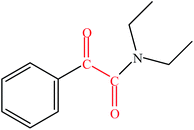 |
92 |
| 3 |
 |
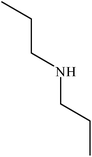 |
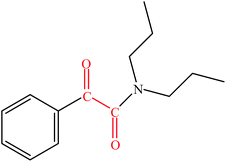 |
87 |
| 4 |
 |
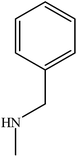 |
 |
89 |
| 5 |
 |
 |
 |
98 |
| 6 |
 |
 |
 |
95 |
| 7 |
 |
 |
 |
96 |
| 8 |
 |
 |
 |
88 |
| 9 |
 |
 |
 |
95 |
| 10 |
 |
 |
 |
94 |
Proposed mechanism. The full mechanistic details of this transformation have not been determined; however a plausible reaction mechanism is shown in Scheme 5 based on previous reports.30 Palladium nano particles are stabilized by nitrogen containing mesoporous mPMF. These Pd nano particles are the active centres which are responsible for both the carbonylation reactions. It is generally accepted that an organopalladium halide is produced by oxidative addition of an organic aryl halide to the Pd(0) species but the later course of the catalytic reaction may vary depending on the nucleophiles and reaction conditions employed. The most often assumed process involves migratory insertion of a coordinated carbon monoxide to give an acylpalladium halide. It is found that when CO pressure is low (3 atm) then the acylpalladium halide species reacts further with a nucleophile (amine) and a base to liberate the amide (mono carbonylative product). But in the presence of high CO pressure (20 atm) acylpalladium halide species further coordinates with CO and then it reacts with amine and base to give α-ketoamide (double carbonylative product).
 |
| | Scheme 5 Proposed reaction mechanism of palladium catalyzed carbonylation of aryl halide. | |
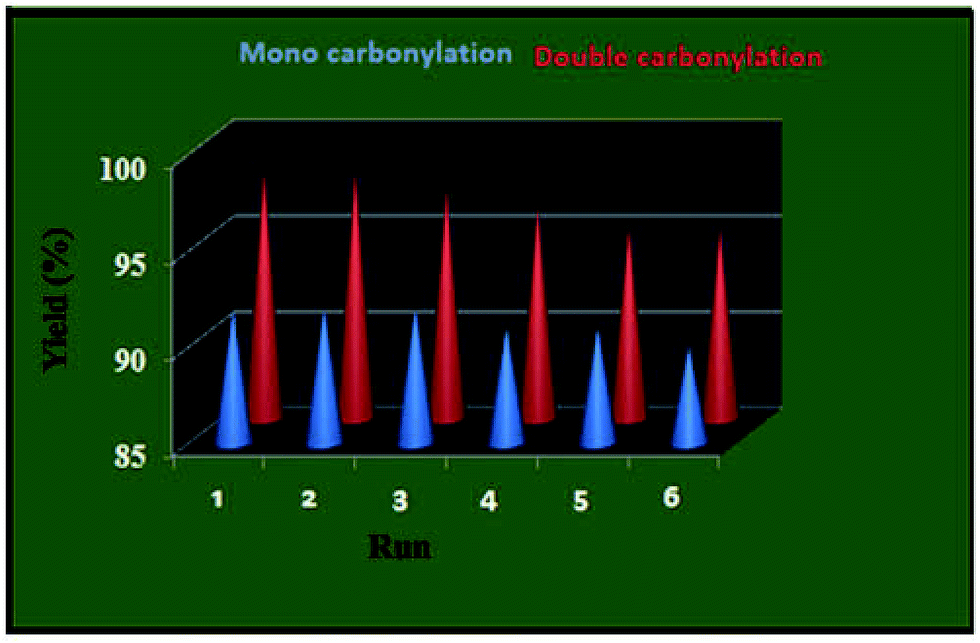
Catalyst reusability. Recovery and catalyst reuse are important issues in the heterogeneous catalysis. We studied the reusability of the present heterogeneous palladium nano catalyst in the mono and double carbonylation of aryl halide with morpholine under the optimized reaction conditions. After completion of the reactions, the catalyst was recovered by simple filtration and washed with ethyl acetate followed by acetone then dried at 40 °C. The recovered catalyst was employed in the next run with further addition of substrates in appropriate amount under optimum reaction conditions. The catalyst show almost same activity up to six reaction cycles (Fig. 8). No catalyst deterioration was observed, confirming the high stability of the heterogeneous catalyst under the reaction conditions.
 |
| | Fig. 8 Catalyst reusability test of the mPMF–Pd0 catalyst. | |
4. Conclusions
In conclusion, we have reported the preparation and characterization of a highly stable, reusable nano catalyst, mPMF–Pd0 and its successful applications for the mono and double amino carbonylations of aryl halides with morpholine, exploiting a simple setup and reaction conditions. Both electron-rich and electron-deficient aryl halides could be applied and the nature of the base, temperature and the solvent system proved crucial for these transformations. The present system is highly air and moisture stable and the catalyst can be synthesized readily from inexpensive and commercially available starting materials. Moreover, the catalyst was reused for six consecutive cycles with consistent catalytic activity. Further work is in progress to broaden the scope of this catalytic system for other organic transformation. A protocol, which allows expansion of this chemistry to other aromatic halides and exact mechanistic pathway of this reaction are currently under investigation.
Acknowledgements
SMI acknowledges CSIR and DST, New Delhi for financial support. RAM acknowledges UGC, New Delhi, India for his Maulana Azad National Fellowship (F1-17.1/2012-13/MANF-2012-13-MUS-WES-9628/SA-III). ASR acknowledges CSIR, New Delhi, India for his senior research fellowship.
References
-
(a) A. Roucoux, J. Schultz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed;
(b) M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed;
(c) J. Baca, J. H. Ahn, Y. Sun, M. A. Meitl, E. Menard, H. S. Kim, W. M. Choi, D. H. Kim, Y. Huang and J. A. Rogers, Angew. Chem., Int. Ed., 2008, 47, 5524–5542 CrossRef PubMed.
-
(a) H. Hirai, N. Yakura, Y. Seta and S. Hodoshima, React. Funct. Polym., 1998, 37, 121–131 CrossRef CAS;
(b) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
-
(a) R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed;
(b) A. Prastaro, P. Ceci, E. Chiancone, A. Boffi, R. Cirilli, R. M. Colone, G. Fabrizi, A. Stringaro and S. Cacchi, Green Chem., 2009, 11, 1929–1932 RSC.
-
(a) S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4, 1147–1151 CrossRef CAS;
(b) D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS PubMed;
(c) Z. Zhang and Z. Wang, J. Org. Chem., 2006, 71, 7485–7487 CrossRef CAS PubMed.
-
(a) N. Yan, J. g. Zhang, Y. Tong, S. Yao, C. Xiao, Z. Li and Y. Kou, Chem. Commun., 2009, 4423–4425 RSC;
(b) Y. Hu, Y. Yu, Z. Hou, H. Li, X. Zhao and B. Feng, Adv. Synth. Catal., 2008, 350, 2077–2085 CrossRef CAS.
- A. Sachse, N. Linares, P. Barbaro, F. Fajulaa and A. Galarneau, Dalton Trans., 2013, 1378–1384 RSC.
- R. Xing, Y. Liu, H. Wu, X. Li, M. He and P. Wu, Chem. Commun., 2008, 6297–6299 RSC.
- M. Faticanti, N. Cioffi, S. D. Rossi, N. Ditaranto, P. Porta, L. Sabbatini and T. B. Zacheo, appl. Catal. B, 2005, 60, 73–82 CrossRef CAS PubMed.
-
(a) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed;
(b) J. A. Melero, R. V. Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed;
(c) S. Angelos, E. Johansson, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2261–2271 CrossRef CAS;
(d) M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef PubMed;
(e) A. Taguchi and F. Suchuth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS PubMed.
-
(a) F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS PubMed;
(b) Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
-
(a) N. Salam, S. K. Kundu, A. S. Roy, P. Mondal, S. Roy, A. bhaumik and S. M. Islam, Catal. Sci. Technol., 2013, 3, 3303–3316 RSC;
(b) U. Mandi, M. Pramanik, A. S. Roy, N. Salam, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 15431–15440 RSC;
(c) N. Salam, B. Banerjee, A. S. Roy, P. Mondal, S. Roy, A. bhaumik and S. M. Islam, Appl. Catal., A, 2014, 477, 184–194 CrossRef CAS PubMed.
-
(a) Y. Meng, D. Gu, F. Zhang, Y. F. Shi, L. Cheng, D. Feng, Z. X. Wu, Z. X. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS;
(b) B. Karimi, H. M. Mirzaei and A. Mobaraki, Catal. Sci. Technol., 2012, 2, 828–834 RSC.
- D. Chandra, B. K. Jena, C. R. Raj and A. Bhaumik, Chem. Mater., 2007, 19, 6290–6296 CrossRef CAS.
-
(a) M. Nandi, R. Gangopadhyay and A. Bhaumik, Microporous Mesoporous Mater., 2008, 109, 239–247 CrossRef CAS PubMed;
(b) M. Pramanik, M. Nandi, H. Uyama and A. Bhaumik, Catal. Sci. Technol., 2012, 2, 613–620 RSC.
- J. Schuster, R. Koehn, A. Keilbach, M. Doeblinger, H. Amenitsch and T. Bein, Chem. Mater., 2009, 21, 5754–5762 CrossRef CAS.
-
(a) Y. L. Zhang, S. Wei, F. J. Liu, Y. C. Du, S. Liu, Y. Y. Ji, T. Yokoi, T. Tatsumi and F. S. Xiao, Nano Today, 2009, 4, 135–142 CrossRef CAS PubMed;
(b) E. Poli, R. De Sousa, F. Jerome, Y. Pouilloux and J. M. Clacens, Catal. Sci. Technol., 2012, 2, 910–914 RSC.
- J. K. Cho, R. Najman, T. W. Dean, O. Ichihara, C. Muller and M. Bradley, J. Am. Chem. Soc., 2006, 128, 6276–6277 CrossRef CAS PubMed.
-
(a) Comprehensive Organometallic Chemistry II, ed. B. C. Soderberg, L. S. Hegedus, E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, 1995, vol. 12, pp. 249–251 Search PubMed;
(b) J. Tsuji, Palladium Reagents and Catalysis, Wiley, Chichester, 1995 Search PubMed;
(c) R. F. Heck, Palladium Reagents in Organic Syntheses, Academic Press, London, 1985 Search PubMed.
-
(a) N. A. Meanwell, O. B. Wallace, H. Fang, H. Wang, M. Deshpande, T. Wang, Z. Yin, Z. Zhang, B. C. Pearce, J. James, K. S. Yeung, Z. Qiu, J. J. K. Wright, Z. Yang, L. Zadjura, D. L. Tweedie, S. Yeola, F. Zhao, S. Ranadive, B. A. Robinson, Y. F. Gong, H. G. H. Wang, W. S. Blair, P. Y. Shi, R. J. Colonno and P. F. Lin, Bioorg. Med. Chem. Lett., 2009, 19, 1977–1984 CrossRef CAS PubMed;
(b) S. K. V. Vernekar, H. Y. Hallaq, G. Clarkson, A. J. Thompson, L. Silvestri, S. C. R. Lummis and M. Lochner, J. Med. Chem., 2010, 53, 2324–2328 CrossRef CAS PubMed;
(c) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6946–6948 CrossRef CAS PubMed.
-
(a) F. X. Tavares, D. N. Deaton, A. B. Miller, L. R. Miller, L. L. Wright and H. Q. Zhou, J. Med. Chem., 2004, 47, 5049–5056 CrossRef CAS PubMed;
(b) S. D. Lewis, B. J. Lucas, S. F. Brady, J. T. Sisko, K. J. Cutrona, P. E. J. Sanderson, R. M. Freidinger, S. S. Mao, S. J. Gardell and J. A. Shafer, J. Biol. Chem., 1998, 273, 4853–4854 Search PubMed;
(c) B. A. Malcolm, R. Liu, F. Lahser, S. Agrawal, B. Belanger, N. Butkiewicz, R. Chase, F. Gheyas, A. Hart, D. Hesk, P. Ingravallo, C. Jiang, R. Kong, J. Lu, J. Pichardo, A. Prongay, A. Skelton, X. Tong, S. Venkatraman, E. Xia, V. Girijavallabhan and F. G. Njoroge, Antimicrob. Agents Chemother., 2006, 50, 1013–1020 CrossRef CAS PubMed;
(d) T. Wang, Z. Zhang, O. B. Wallace, M. Deshpande, H. Fang, Z. Yang, L. M. Zadjura, D. L. Tweedie, S. Huang, F. Zhao, S. Ranadive, B. S. Robinson, Y. F. Gong, K. Ricarrdi, T. P. Spicer, C. Deminie, R. Rose, H. G. H. Wang, W. S. Blair, P. Y. Shi, P. F. Lin, R. J. Colonno and N. A. Meanwell, J. Med. Chem., 2003, 46, 4236–4239 CrossRef CAS PubMed;
(e) Q. Guo, H. T. Ho, I. Dicker, L. Fan, N. Zhou, J. Friborg, T. Wang, B. V. McAuliffe, H. g. H. Wang, R. E. Rose, H. Fang, H. T. Scarnati, D. R. Langley, N. A. Meanwell, R. Abraham, R. J. Colonno and P. f. Lin, J. Virol., 2003, 77, 10528–10536 CrossRef CAS.
-
(a) C. A. Montalbetti and G. N. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS PubMed;
(b) R. Vaidyanathan, V. G. Kalthod, D. P. Ngo, J. M. Manley and S. P. Lapekas, J. Org. Chem., 2004, 69, 2565–2568 CrossRef CAS PubMed;
(c) R. C. Larock, Comprehensive Organic Transformation, VCH, New York, 1999 Search PubMed;
(d) D. M. Shendage, R. Freoehlich and G. Haufe, Org. Lett., 2004, 6, 3675–3678 CrossRef CAS PubMed;
(e) J. C. Baum, J. E. Milne, J. A. Murry and O. R. Thiel, J. Org. Chem., 2009, 74, 2207–2209 CrossRef CAS PubMed;
(f) L. D. S. Yadav and V. P. S. Garima, Tetrahedron Lett., 2010, 51, 739–742 CrossRef CAS PubMed.
-
(a) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS PubMed;
(b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS;
(c) Y. Uozumi, T. Arii and T. Watanabe, J. Org. Chem., 2001, 66, 5272–5274 CrossRef CAS;
(d) M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287–293 CrossRef CAS;
(e) S. Ho, G. Bondarenko, D. Rosa, B. Dragisic and A. Orellana, J. Org. Chem., 2012, 77, 2008–2012 CrossRef CAS PubMed;
(f) V. Fuente, C. Godard, C. Claver and S. Castillon, Adv. Synth. Catal., 2012, 354, 1971–1979 CrossRef;
(g) Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–155 CrossRef CAS PubMed;
(h) M. Papp and R. S. Földes, J. Mol. Catal. A: Chem., 2013, 378, 193–199 CrossRef CAS PubMed.
-
(a) J. Salvadori, E. Balducci, S. Zaza, E. Petricci and M. Taddei, J. Org. Chem., 2010, 75, 1841–1847 CrossRef CAS PubMed;
(b) T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805–1807 RSC;
(c) T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406–1410 CrossRef CAS;
(d) J. Liu, S. Zheng, W. Sun and C. Xia, Chin. J. Chem., 2009, 27, 623–627 CrossRef CAS;
(e) J. Balogh, A. Kuik, L. Urge, F. Darvas, J. Bakos and R. Skoda-Foldes, J. Mol. Catal. A: Chem., 2009, 302, 76–79 CrossRef CAS PubMed.
- M. X. Tan, L. Gu, N. Li, J. Y. Ying and Y. Zhang, Green Chem., 2013, 15, 1127–1132 RSC.
-
(a) P. Pandey, A. P. Katsoulidis, I. Eryazici, Y. Wu, M. G. Kanatzidis and S. T. Nguyen, Chem. Mater., 2010, 22, 4974–4979 CrossRef CAS;
(b) J. Germain, F. Svec and J. M. J. Frechet, Chem. Mater., 2008, 20, 7069–7076 CrossRef CAS;
(c) P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS;
(d) M. G. Schwab, B. Fassbender, H. W. Spiess, A. Thomas, X. Feng and K. Mullen, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- M. N. Al-Hinai, R. Hassanien, N. G. Wright, A. B. Horsfall, A. Houlton and B. R. Horrocks, Faraday Discuss., 2013, 164, 71–91 RSC.
-
(a) A. K. Thapaa, B. Pandita, H. Sharma Paudel, R. Thapa, S. Ida, J. Jasinskia, G. U. Sumanasekeraa and T. Ishihara, Electrochim. Acta, 2014, 127, 410–415 CrossRef PubMed;
(b) A. C. Ferrari and J. Robertson, Phys. Rev. B: Solid State, 2001, 64, 075414 CrossRef.
- Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and G. An, J. Colloid Interface Sci., 2006, 304, 323–328 CrossRef CAS PubMed.
- M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaudz, Catal. Sci. Technol., 2012, 2, 1886–1893 CAS.
-
(a) Y. Uozumi, T. Arii and T. Watanabe, J. Org. Chem., 2001, 66, 5272–5274 CrossRef CAS;
(b) N. Tsukada, Y. Ohba and Y. Inoue, J. Organomet. Chem., 2003, 687, 436–443 CrossRef CAS PubMed;
(c) R. E. Rulke, V. E. Kaasjager, P. Wehman, C. J. Elsevier, P. W. N. M. van Leeuwen and K. Vrieze, Organometallics, 1996, 15, 3022–3031 CrossRef;
(d) Y. Lin and A. Yamamoto, Organometallics, 1998, 17, 3466–3478 CrossRef CAS;
(e) F. Christopher and J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef;
(f) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4143 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07554d |
| ‡ These authors contributed equally to this manuscript. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.