
Carbon-coated Ni3Sn2 nanoparticles embedded in porous carbon nanosheets as a lithium ion battery anode with outstanding cycling stability†
Jian Qina,
Xiang Zhanga,
Naiqin Zhaoab,
Chunsheng Shia,
Enzuo Liua,
Jiajun Lia and
Chunnian He*ab
aSchool of Materials Science and Engineering and Tianjin Key Laboratory of Composites and Functional Materials, Tianjin University, Tianjin 300072, China
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300072, China. E-mail: cnhe08@tju.edu.cn
First published on 16th September 2014
Abstract
Carbon-coated Ni3Sn2 nanoparticles uniformly embedded in two-dimensional porous carbon nanosheets (2D Ni3Sn2@C@PGC) as superior lithium ion battery anode material were fabricated by a facile and scalable method, which involves in situ synthesis of 2D Ni@C@PGC and chemical vapor transformation processes from 2D Ni@C@PGC to Ni3Sn2@C@PGC. With the assistance of a water-soluble cubic NaCl template, 2D Ni@C@PGC was firstly in situ synthesized on the surface of NaCl particles. After vapor transformation with SnCl2, the Ni@C@PGC nanosheets were converted to Ni3Sn2@C@PGC, in which uniform Ni3Sn2 nanoparticles coated with conformal graphitized carbon layers were homogeneously embedded in 2D high-conducting carbon nanosheets with a thickness of about 30 nm. This unique 2D dual encapsulation structure with high porosity, high electronic conductivity, outstanding mechanical flexibility and short lithium ion diffusion pathway is favorable for lithium insertion and extraction during deep charge–discharge processes. As a result, the electrode fabricated using 2D Ni3Sn2@C@PGC as the anode and a lithium plate as the cathode exhibits a high reversible capacity up to 585.3 mA h g−1 at a current density of 0.2 C (1 C = 570 mA h g−1) after 100 cycles, a high rate capability (484, 424, 378, 314 and 188 mA h g−1 at 0.2, 0.5, 1, 2 and 5 C, respectively, 1 C = 570 mA h g−1), and superior cycling stability at a high rate (350.3 mA h g−1 at a rate of 1 C after 180 cycles).
1. Introduction
Rechargeable lithium ion batteries (LIBs) are one of the most common energy storage devices, with applications from mobile phones to ongoing research for hybrid vehicles.1,2 Graphite is used in commercial anode materials because of its low cost, good conductivity and electrochemical stability. However, with limited theoretical capacity of 372 mA h g−1 (LiC6), graphite can no longer meet demand for LIBs with high energy density and power density. Therefore, there is extensive interest in novel high-capacity anode materials. Lithium alloys (LAs), such as Sn, Ge, Sb, etc., which have large theoretical capacity and high electrical conductivity,3–10 have been explored extensively as alternative anode materials for high-performance LIBs. For example, metallic Sn reversibly transforms between Li4.4Sn and Sn during lithiation/delithiation processes, giving a theoretical specific capacity of 992 mA h g−1 (Li4.4Sn) based on calculation.11 However, the main obstacle to developing LA-based anodes is their dramatic volume change associated with the lithium ion insertion/extraction processes, which would result in their pulverization and exfoliation from the current collector, thereby leading to poor cycling stability.12,13Several approaches have been developed aiming at overcoming this obstacle. The first route, nanostructuring, relies on creation of short diffusion paths for lithium ion transport and free space to accommodate the large volume change.14–18 Although the nanostructured materials can accommodate the mechanical strain of lithium ion insertion/extraction much better than can the bulk materials, the limited structural stability of the nanostructured LAs remains problematic. Without protection, bare LA nanostructures cannot maintain their original morphology because of pulverization resulting from the repeated lithiation/delithiation. The second approach is to synthesize active/inactive M1M2 intermetallics, in which the electrochemically active metal M1 (Sn, Ge, Sb) forms the desired lithium alloy, LiM1, with the other metal M2 (Co, Fe, Ni, etc.) acting as an electrochemically inactive matrix to absorb the massive volume changes occurring during the lithiation/delithiation processes as well as serving as an electrical conductor to enhance electrical integration in the anode.19–23 The large volume change of LAs on lithium insertion can be accommodated to a limited extent by these buffer matrices; however, internal stress in active materials induced by volume expansion cannot be effectively eliminated, resulting in the electrodes suffering from serious stress-induced cracking and, in turn, in capacity fading after prolonged cycling.24,25 The third approach, constructing hybrid electrodes composed of LAs and nanostructured carbon with specific structure, aims at improving the structural stability through use of carbon as a protective coating layer or matrix.6–10 The carbon layer or matrix serves three main purposes: (1) to wire the active LAs, (2) to buffer the volume change and prevent the aggregation of pulverized active material, and (3) to protect the LAs against direct contact with the electrolyte, which may result in formation of an unstable solid electrolyte interphase (SEI) film. However, because this design does not provide appropriate space to accommodate the large volume expansion, the carbon layer may fracture on repeated lithiation/delithiation, leading to electrical disconnection, re-exposure of the active materials to electrolyte, and active material aggregation. In addition, formulation of a uniform carbon coating on each metal surface is impossible in the molecular dimension, which poses problems.
Recently, great attention has been directed towards two-dimensional (2D) nanostructures because of their fascinating physical and chemical properties.26–30 In particular, development of graphene and transitional metal oxide nanostructures has been pursued intensively on the basis of fundamental scientific interest coupled with wide potential for many technological applications. Generally speaking, a 2D nanostructure exhibits unique mechanical, electrical, and optical properties because of its large surface area and miniscule atomic scale thicknesses. In this context, 2D nanostructures used as LIB electrodes could exhibit much improved kinetics because of a shortened pathway for lithium ion diffusion and large exposed surface offering more lithium-insertion channels.31–33
In this work, we use the advantages of nanostructuring, alloying, core/shell encapsulation structures and 2D nanostructured design for preparation of discrete, homogeneous and ultrasmall carbon-encapsulated Ni3Sn2 nanoparticles embedded in 2D porous graphitic carbon nanosheets (indicated with 2D Ni3Sn2@C@PGC nanosheets) as superior LIB anodes. The novel strategy involves in situ preparation of carbon-encapsulated Ni nanoparticles embedded in porous graphitic carbon nanosheets (Ni@C@PGC) with the assistance of the catalytic ability of Ni particles and the surface of NaCl template,10 and substitution of partial Ni with Sn to form Ni3Sn2 (inactive/active intermetallic) nanoparticles by a chemical vapor transformation (CVT) method.34–36 In this unique dual encapsulated structure, that is graphitic onion-like carbon-coated Ni3Sn2 nanoparticles encapsulated in 2D porous carbon nanosheets, the onion-like carbon shells can effectively prevent Ni3Sn2 nanoparticles from aggregation as well as direct exposure to electrolyte, which is favorable for maintaining the structural and interfacial stabilization of Ni3Sn2 nanoparticles. Meanwhile, the flexible and conductive graphitic carbon nanosheets with high porosity can not only accommodate the mechanical stress caused by volume change of Ni3Sn2 nanoparticles, but also provide high surface area and short diffusion pathway for transfer of lithium ions and electrons, which are beneficial for maintaining structural and electrical integrity of the overall electrode during the charge/discharge processes.10 As a result, this novel 2D Ni3Sn2@C@PGC nanosheet electrode exhibits tremendously improved lithium storage performance compared with previous NiSn-based anode materials and NiSn/carbon hybrid anodes. It shows a high reversible capacity up to 585.3 mA h g−1 at a current density of 0.2 C (1 C = 570 mA h g−1) after 100 cycles, a high rate capability (484, 424, 378, 314 and 188 mA h g−1 at 0.2, 0.5, 1, 2 and 5 C, respectively, 1 C = 570 mA h g−1), and superior cycling performance at a high rate (350.3 mA h g−1 at a rate of 1 C after 180 cycles).
2. Experimental
2.1 Synthesis of 2D Ni3Sn2@C@PGC nanosheets
Metal precursors of Ni(NO3)2·6H2O (0.99 g), citric acid (2.5 g), and sodium chloride (NaCl) (15 g) were dissolved in 50 mL of deionized water. The resulting mixed solution was dried in a drying oven at 80 °C for 24 h and then ground by agate mortar to obtain very fine composite powders. After that, the composite powders (10 g) together with SnCl2·2H2O powders (1 g) were placed 1 cm apart in a tube furnace and heated at a speed of 5 °C min−1 to 750 °C, and then kept at 750 °C for 2 h under Ar (100 mL min−1). Once cooled to room temperature, the as-synthesized product was washed with deionized water several times to dissolve the NaCl and Ni salt, and then pure 2D Ni3Sn2@C@PGC nanosheets were obtained. For comparison, Ni3Sn2/C nanoblocks were also synthesized by the same procedures without using NaCl.2.2 Characterization techniques
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were performed on a FEI Tecnai G2 F20 TEM. A Raman spectrum was recorded on the LabRAM HR Raman spectrometer using laser excitation at 514.5 nm from an argon ion laser source. X-ray diffraction (XRD) measurements were taken on a Rigaku D/max diffractometer with Cu KR radiation. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer (TA Instruments) up to 800 °C at a heating rate of 10 °C min−1 in air. Brunauer–Emmett–Teller (BET) surface areas and porosities of the products were determined by nitrogen adsorption and desorption using a Micromeritics ASAP 2020 analyzer.2.3 Electrochemical measurements
The working electrodes were made as follows: active materials (2D Ni3Sn2@C@PGC nanosheets, Ni3Sn2/C nanoblocks), conductivity agent (carbon black), and binder (polyvinylidene fluoride, PVDF) in a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 were blended with N-methylpyrrolidone as solvent. Electrode film prepared by coating the mixture on a copper foil was first vacuum-dried at 80 °C for 0.5 h and then at 120 °C for 12 h. Coin cells (CR2032) were fabricated using lithium metal as the counter electrode, Celgard 2400 as the separator, and LiPF6 (1 M) in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%) as the electrolyte. The assembly of the cell was conducted in an Ar-filled glove box followed by an overnight aging treatment before the test. Cyclic voltammetry (CV) measurement was conducted at 0.1 mV s−1 within the range of 0.05–3.0 V on a CHI660D electrochemical workstation. The cycling performance and rate capability of the cells were tested within a fixed voltage window of 0.005–3.00 V (vs. Li+/Li) using a battery testing system (LAND CT 2001A, China) at room temperature. All of the specific capacities here were calculated on the basis of the total weight of 2D Ni3Sn2@C@PGC nanosheets, Ni3Sn2/C nanoblocks. Electrochemical impedance spectroscopy (EIS) measurements were performed using a CHI660D electrochemical workstation employing an AC voltage of 5 mV amplitude in the frequency range 0.1–100 kHz.39
:10:10 were blended with N-methylpyrrolidone as solvent. Electrode film prepared by coating the mixture on a copper foil was first vacuum-dried at 80 °C for 0.5 h and then at 120 °C for 12 h. Coin cells (CR2032) were fabricated using lithium metal as the counter electrode, Celgard 2400 as the separator, and LiPF6 (1 M) in ethylene carbonate/dimethyl carbonate/diethyl carbonate (EC/DMC/DEC, 1:1:1 vol%) as the electrolyte. The assembly of the cell was conducted in an Ar-filled glove box followed by an overnight aging treatment before the test. Cyclic voltammetry (CV) measurement was conducted at 0.1 mV s−1 within the range of 0.05–3.0 V on a CHI660D electrochemical workstation. The cycling performance and rate capability of the cells were tested within a fixed voltage window of 0.005–3.00 V (vs. Li+/Li) using a battery testing system (LAND CT 2001A, China) at room temperature. All of the specific capacities here were calculated on the basis of the total weight of 2D Ni3Sn2@C@PGC nanosheets, Ni3Sn2/C nanoblocks. Electrochemical impedance spectroscopy (EIS) measurements were performed using a CHI660D electrochemical workstation employing an AC voltage of 5 mV amplitude in the frequency range 0.1–100 kHz.39
3. Results and discussion
3.1 Structural and morphological results
Our approach for fabricating 2D Ni3Sn2@C@PGC nanosheets consists of only one step as illustrated in Fig. 1. In a typical synthesis, NaCl, Ni(NO3)2·6H2O and citric acid were mixed to form a homogeneous aqueous solution, and the resulting mixed solution was dried and then ground to obtain very fine composite powders.10 During this process, the NaCl particles were uniformly coated with an ultrathin Ni(NO3)2·6H2O–citric acid complex (as shown in Fig. S1a†), which was induced by chelation between metal ions and the functional groups of citric acid. After that, the composite powders together with SnCl2·2H2O powders (put the two powders into the tube furnace 1 cm apart, not mixture) were calcined under Ar. During the calcining process at lower temperature (<600 °C), the metal precursor (Ni(NO3)2·6H2O) is decomposed to nickel oxide, and the carbon species from C6H12O6 can reduce nickel oxide to Ni nanoparticles,37,38 which can catalyze the carbon from C6H12O6 to form an encapsulating carbon layer around Ni nanoparticles or porous graphitic carbon. As a result, the coating layer on the surface of the NaCl particles was converted to carbon-encapsulated Ni nanoparticles embedded in a porous graphitic carbon nanosheet with uniform thickness (Fig. S1b†). When the calcining temperature was increased above the evaporating point (623 °C) of SnCl2, the SnCl2 vapor could be carried by Ar into the Ni@C@PGC nanosheets to exchange with Ni, and then 2D Ni3Sn2@C@PGC nanosheets were obtained. To verify this process, we also performed the above experiments at 600 °C (lower than the evaporating point of SnCl2) and found that only Ni@C@PGC nanosheets were obtained, as shown in Fig. S2,† further indicating that Ni@C@PGC nanosheets have not exchanged with SnCl2 during the calcining process at a lower temperature. | ||
| Fig. 1 Schematic illustration of the in situ CVT process for one-step synthesis of 2D Ni3Sn2@C@PGC nanosheets using the surface of NaCl as a template. | ||
The crystalline structure of the 2D Ni3Sn2@C@PGC was investigated by XRD (as shown in Fig. 2a). Most of the diffraction peaks, especially those with high intensity, can be assigned to Ni3Sn2 (JCPDS 06-0414), while a small diffraction peak at 26.2° from graphitic carbon also can be observed, suggesting a total conversion from pre-products (Ni@C@PGC nanosheets) to the target sample (2D Ni3Sn2@C@PGC nanosheets). The sharpness of the diffraction peaks also implies that the metallic phase in the products is well-crystallized. In addition, no diffraction peaks from NaCl and Ni salt can be detected, indicating that the NaCl and Ni salt can be removed completely by water washing. The chemical composition of 2D Ni3Sn2@C@PGC nanosheets was investigated by TGA in air at a heating rate of 10 °C min−1 and the result is shown in Fig. 2b. The sample was heated up to 800 °C so that Ni3Sn2 was oxidized to NiO and SnO2 completely and carbon was oxidized to CO2 (Fig. S3†). According to the final content of metal oxide, the original content of Ni3Sn2 was calculated to be 60.07% by weight.
 | ||
| Fig. 2 (a) XRD pattern, (b) TGA profile and (c–e) SEM images of 2D Ni3Sn2@C@PGC nanosheets. | ||
The morphology and microstructure of 2D Ni3Sn2@C@PGC nanosheets were investigated by SEM and TEM. Representative SEM images of the as-synthesized Ni3Sn2@C@PGC sample are shown in Fig. 2c–e. Low-magnification SEM images (Fig. 2c and d) indicate that the products have a predominantly 2D nanosheet and porous morphology, in which Ni3Sn2 nanoparticles are homogeneously embedded. A high-magnification SEM image (Fig. 2e) shows that the thickness of 2D Ni3Sn2@C@PGC nanosheets is about 30 nm. However, when we performed calcination experiments of the composite powders without NaCl, 3D nanoblocks with large Ni3Sn2 particles embedded were obtained (as shown in Fig. S4†), which demonstrates that the surface of water-soluble cubic NaCl can be used effectively as a template for synthesizing 2D nanostructures.10,39–41 In addition, influence of the amount of NaCl in precursor on 2D nanosheet structures has been investigated, and the results are shown in Fig. S5.† Relatively thicker nanosheets and much larger Ni3Sn2 particles with severe aggregation embedded in the nanosheets were obtained at half the amount of NaCl in precursor, indicating that the thickness of 2D nanosheets can be adjusted by controlling the amount of NaCl in precursor.42,43 Fig. 3a and b shows the TEM images of 2D Ni3Sn2@C@PGC nanosheets, with well-dispersed and uniform Ni3Sn2 nanoparticles (5–30 nm) with an almost spherical shape, homogeneously embedded in 2D nanosheets. It is also noted that these Ni3Sn2 nanoparticles are not agglomerated at all by the coalescence of the outer shells, although many papers have mentioned that magnetostatic energy of magnetic nanoparticles can be minimized by such coalescence.44,45 Other products such as carbon nanotubes or fibers and carbon nanoparticles without encapsulation of the metal were scarcely observed in this work. Fig. 3c shows a HRTEM image of 2D Ni3Sn2@C@PGC nanosheets, which clearly reveals that the Ni3Sn2 nanoparticles (5–30 nm), highly crystallized and uniformly coated with well-graphitized onion-like carbon shells with a thickness of about 2 nm, are evenly embedded in the 2D porous carbon nanosheets. The well-maintained 2D structure demonstrates that the SnCl2 vapor effectively diffused into the nanosheets and penetrated the outer carbon shells to exchange with Ni core without damaging the structure of 2D nanosheets. EDS (Fig. S6†), EDX (Fig. S7†), and selected area electronic diffraction (SAED) (inset of Fig. 3e) investigations further verify the encapsulated core being Ni3Sn2 nanoparticles, in perfect accordance with the above XRD results.
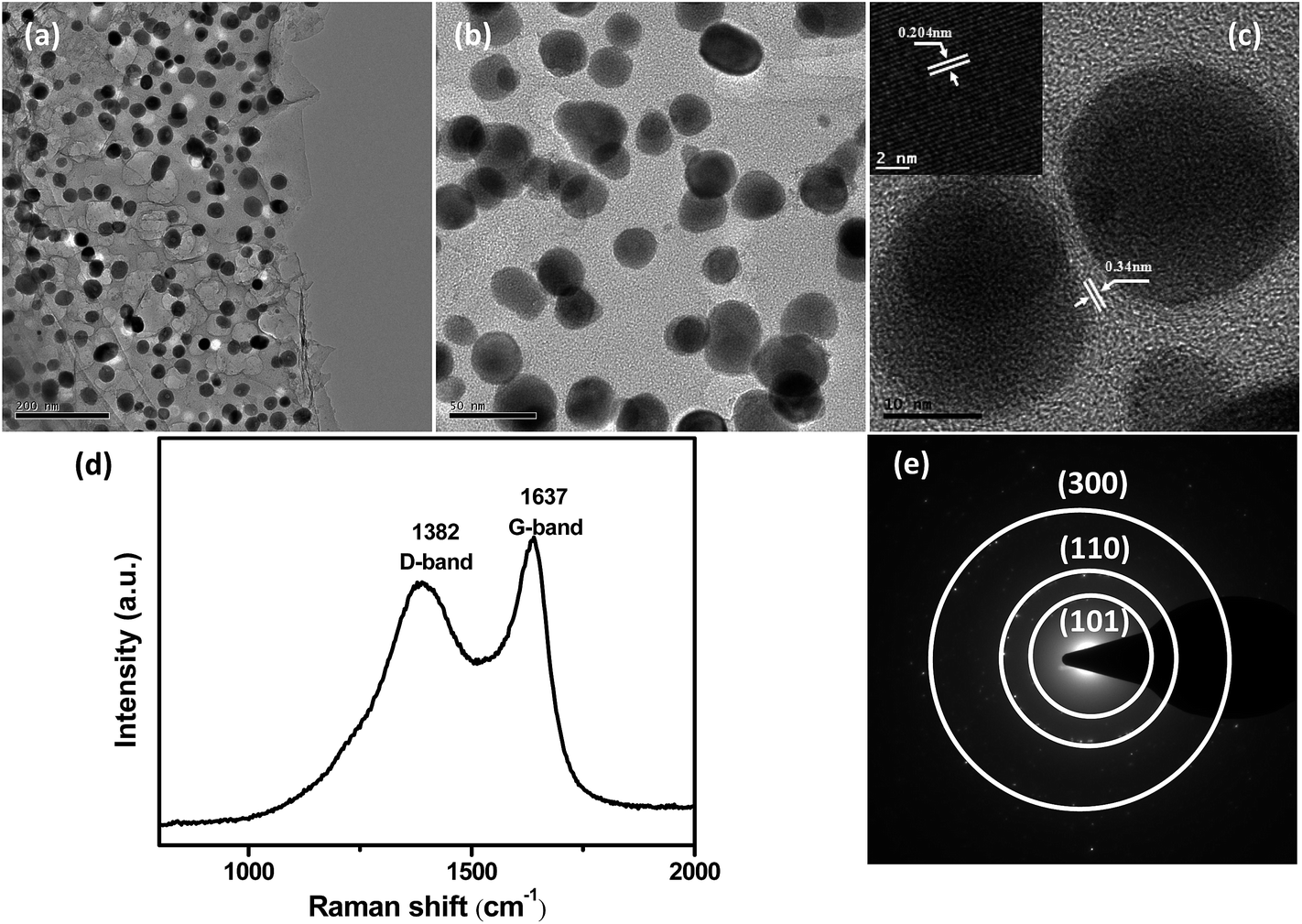 | ||
| Fig. 3 (a and b) TEM images, (c) HRTEM image of Ni3Sn2@C@PGC nanosheet. (d) Raman spectrum and (e) diffraction pattern of Ni3Sn2@C@PGC nanosheet. | ||
The 2D Ni3Sn2@C@PGC nanosheets were also characterized by Raman spectroscopy in detail to further estimate the ordering degree of carbon structure in the nanosheets. A typical Raman spectrum collected within the 800–2000 cm−1 range is shown in Fig. 3d. It can be seen that the spectrum exhibits two obvious peaks, corresponding to the D band (1382 cm−1) and G band (1637 cm−1). The former band (D band) is associated with edges, defects, and disordered carbon, whereas the latter band (G band) is ascribed to sp2-hybridized carbon.46 The intensity ratio of G peak to D peak (IG/ID) is widely used to assess crystallization of carbon materials. The IG/ID ratio of our 2D Ni3Sn2@C@PGC nanosheets was calculated to be ∼1.16, implying that the carbon in the nanosheets we obtained is well-crystallized graphitic carbon, which would be beneficial for enhancing the electronic conduction of the overall electrode. Nitrogen adsorption–desorption measurements were carried out at 77 K to study the textural characteristics of the 2D Ni3Sn2@C@PGC nanosheets. For comparison, the Ni3Sn2/C nanoblocks were also characterized by nitrogen adsorption–desorption measurements. As shown in Fig. 4, the isotherm profile of the 2D Ni3Sn2@C@PGC nanosheets exhibits a typical IV isotherm with a hysteresis loop in the P/P0 range of 0.5–0.9, indicating that the pores inside the sample consist of micropores and mesopores, as further verified by the pore size distribution shown in Fig. 4d. The BET specific surface area of our 2D Ni3Sn2@C@PGC nanosheets is measured to be 266 m2 g−1, much higher than that of Ni3Sn2/C nanoblocks (about 51.5 m2 g−1) (see Fig. S8†). The pore size distribution (as shown in Fig. 4b) of the 2D Ni3Sn2@C@PGC nanosheets analyzed by DFT method lies in the 0.5–50 nm range, which includes micropores and mesopores. The pore volume of the product determined by the Barrett–Joyner–Halenda method is 0.194 cm3 g−1. This porous structure consisting of micropores and mesopores with size in the range 0.5–50 nm may be attributed to escape of small molecules in citric acid during calcining, and is likely to aid electrolyte ion diffusion to active sites with less resistance and to tolerate the volume change of the NiSn nanocrystals during charge/discharge cycles.10,39
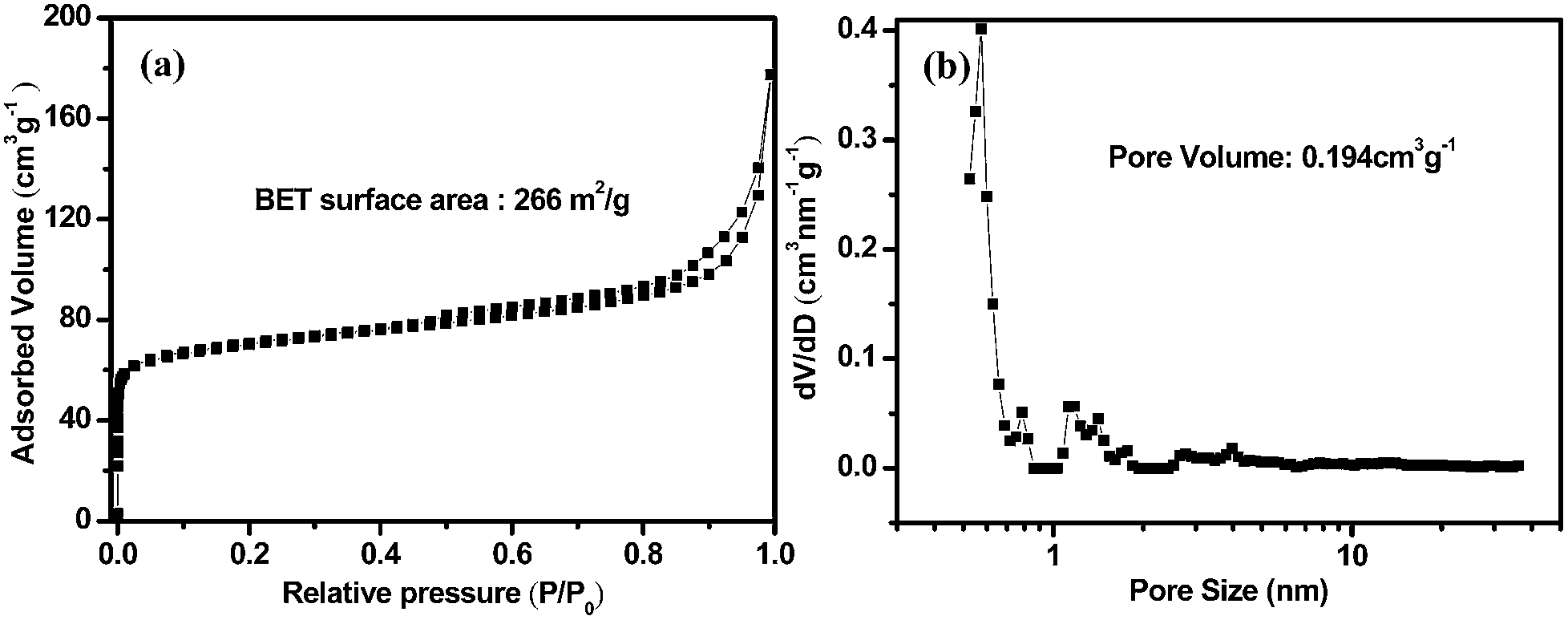 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms and (b) pore distribution analysis by DFT method of Ni3Sn2@C@PGC nanosheet. | ||
3.2 Electrochemical performance of the 2D Ni3Sn2@C@PGC nanosheets
Lithium insertion/extraction reactions of the 2D Ni3Sn2@C@PGC nanosheets were first investigated by CV experiment as shown in Fig. 5a. The test was carried out at a scanning rate of 0.1 mV s−1 within a voltage window of 0.005–3 V (vs. Li/Li+). In the first scanning cycle, two cathodic peaks between 0.5–1 V and 1.5–1.75 V were observed, but disappeared completely in the second cycle. This can be attributed to some irreversible reactions associated with formation of SEI film as well as decomposition of electrolyte. The cathodic peak between 0 and 0.5 V, and anodic peaks at 0.68, 1.3 and 2.1 V correspond to the electrochemical alloying/dealloying reactions. The whole reaction is expected to evolve like this:| Ni3Sn2 + 8.8Li+ + 8.8e− → 2Li4.4Sn + 3Ni | (1) |
| Li4.4Sn → Sn + 4.4Li+ + 4.4e− (charge) | (2) |
| Sn + 4.4Li+ + 4.4e− → Li4.4Sn (discharge) | (3) |
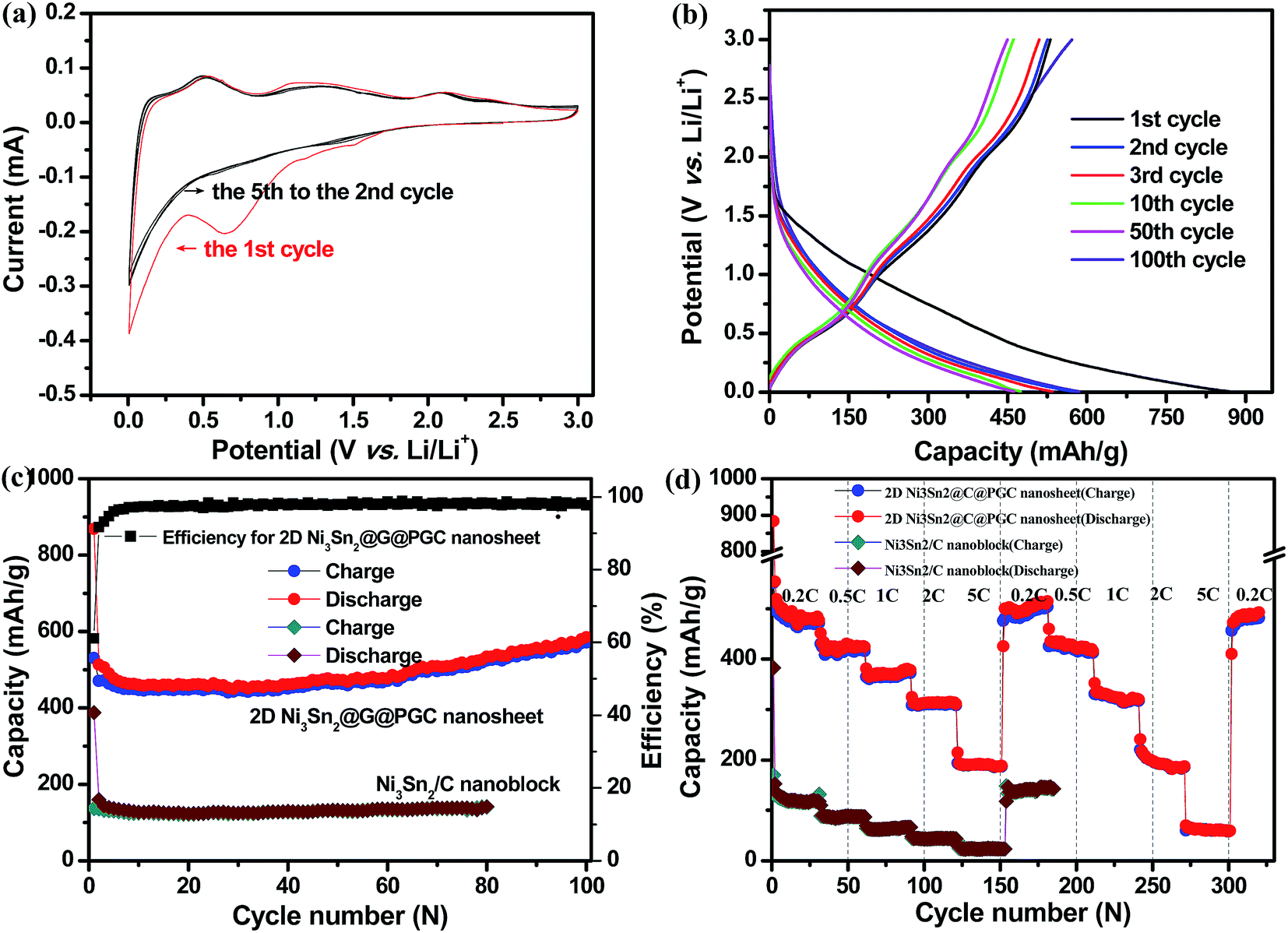 | ||
| Fig. 5 (a) CV curves of the 2D Ni3Sn2@C@PGC electrode at a voltage range of 0.005 to 3.0 V and scan rate of 0.1 mV s−1. (b) Voltage profiles of the 2D Ni3Sn2@C@PGC electrode at a current density of 0.2 C. (c) Cycle performance of the 2D Ni3Sn2@C@PGC and Ni3Sn2/C nanoblocks at a current density of 0.2 C (1 C = 570 mA h g−1). (d) Rate cycle performance of the electrodes of 2D Ni3Sn2@C@PGC and Ni3Sn2/C nanoblocks at charge/discharge rates from 0.2 to 5 C for 320 cycles. | ||
Reaction (1) occurs in the first activation cycle (irreversible), whereas reactions (2) and (3) operate alternately in the following cycles. In addition, it should be noted that from the second to the fifth cycle in our results, the profiles almost overlapped, indicating the perfect reversibility of our electrode.
Representative galvanostatic charge–discharge profiles of 2D Ni3Sn2@C@PGC nanosheets at a current density of 0.2 C (1 C = 570 mA h g−1) are shown in Fig. 5b. The first lithiation process delivers an initial discharge capacity of 868.8 mA h g−1, and subsequent delithiation delivers a charge capacity of 531 mA h g−1, resulting in a Coulombic efficiency of 61.1%. The relatively low initial Coulombic efficiency observed in the first cycle may be attributed to the irreversible capacity loss, including inevitable formation of SEI and decomposition of electrolyte, which are common to most anode materials,3–10 and also agree well with the above CV results that the cathodic peaks are present in the first scan but absent afterward. A prelithiation strategy can be used to improve the low Coulombic efficiency in practical application. The Coulombic efficiency increases dramatically to 96% in the fifth cycle and levels off at 98–100% in subsequent cycles, indicating a facile insertion/extraction of lithium ions and efficient transportation of electrons and ions in this nanosheet anode. Importantly, it should be noted that both charge and discharge profiles exhibit little shift from the second to the 100th cycle, further verifying that the 2D Ni3Sn2@C@PGC nanosheet electrodes are stable during cycling.
The cycling performances of 2D Ni3Sn2@C@PGC nanosheets and Ni3Sn2/C nanoblocks produced without using NaCl at a current density of 0.2 C are shown in Fig. 5c. It is evident that the 2D Ni3Sn2@C@PGC nanosheet electrode shows superior cycling stability and exhibits almost complete stable cycling performance from the second cycle onwards. Even after 100 cycles, a capacity of 585.3 mA h g−1 can be maintained, which is 113% of the capacity at the second cycle. The capacity rise of the Ni3Sn2@C@PGC composite is commonly observed for lithium alloy/carbon composite anodes, and normally attributed to the presence of a possible activation process in the electrode.47–49 In sharp contrast, the Ni3Sn2/C nanoblocks electrode delivers first discharge and charge capacities of 388 mA h g−1 and 137 mA h g−1 only, and is stable at only 141.4 mA h g−1 after 80 cycles. Therefore, the 2D Ni3Sn2@C@PGC nanosheet anode demonstrates a remarkably high reversible capacity and superior cycle performance. This may be ascribed largely to the unique 2D dual carbon-encapsulated structure we have built, that is, graphitic onion-like carbon-coated Ni3Sn2 nanoparticles embedded in 2D porous carbon nanosheets. During the charge–discharge process, the onion-like carbon shells around Ni3Sn2 nanoparticles can not only effectively prevent active metal materials from immediate contact with the electrolyte and thus decrease the site reactions occurring on the surface of active metal materials, but also can restrain the agglomeration of Ni3Sn2 nanoparticles to large particles during cycling.10,39 As for the 2D porous carbon nanosheets with high mechanical flexibility, they can relieve the mechanical stress caused by the volume change of Ni3Sn2 nanoparticles and also act as a stable matrix for SEI film formation, which are very helpful for enhancing the cycling stability. In addition, 2D carbon nanosheets with a porous feature and integrative characteristic can lead to a high contact area between the electrode and electrolyte and a much shorter pathway for lithium ion diffusion, which are beneficial for efficient access of electrolyte into the electrode interior to guarantee a rapid lithium ion insertion/extraction reaction. All the advantages discussed above result in high capacities and outstanding cycle stability.
Fig. 5d demonstrates the rate and rate cycle performance of 2D Ni3Sn2@C@PGC nanosheets and Ni3Sn2/C nanoblocks at different current rates (from 0.2 C to 5 C). It can be seen that the 2D Ni3Sn2@C@PGC nanosheets exhibit a durable and stable rate capacity at different charge/discharge rates. In the first rate cycle, the average reversible capacities are 484, 424, 378, 314 and 188 mA h g−1 at rates of 0.2 (step 1), 0.5 (step 2), 1 (step 3), 2 (step 4) and 5 C (step 5), respectively, and the corresponding power and energy densities for different charge/discharge rates are shown in the Ragone plot in Fig. S9.† This excellent rate performance is superior to that of most Sn-based alloy anode materials.23,50–58 When the current rate returns to the initial 0.2 C after 150 cycles, a capacity as high as 500 mA h g−1 is still recoverable and sustainable up to the 180th cycle, which is even higher than the initial capacity of 484 mA h g−1. This indicates that our nanosheet structure is sufficiently stable so that, even when cycled at a high rate of 5 C, charging/discharging processes finished within 8 minutes, and structural integrity was maintained. Moreover, we performed the whole rates again (from 180 cycles to 320 cycles) to further measure rate cycle performance (as shown in Fig. 5d). Within 320 cycles the second stage of rate cycling (from 180th to 320th cycles) shifted only slightly from the first stage of rate cycling (from 1st to 180th cycles), as shown in Table S1.† When returned to 0.2 C after two rate cycling stages, the electrode still delivered a reversible capacity of 497 mA h g−1. The above results indicate that the structure of Ni3Sn2@C@PGC nanosheets remains stable even under high rate cycling. As a comparison, the Ni3Sn2/C nanoblocks electrode produced without using NaCl shows a significantly lower capacity (as shown in Fig. 5d and Table S1†), further verifying the advantages of using the 2D Ni3Sn2@C@PGC nanosheets for lithium storage. Fig. 6a shows the charge–discharge profiles of 2D Ni3Sn2@C@PGC nanosheets at different rates, from which we can see that the curves at high rates are similar to those at low rates, indicating the stable structure of 2D Ni3Sn2@C@PGC nanosheets even at high charge–discharge rates. Fig. 6b shows the cycle performance of our electrode at high rates: the experiment was carried out at current rates of 0.5 C and 1 C (570 mA h g−1) within a voltage window between 0.005 and 3 V. The reversible capacity at 0.5 C after 250 cycles is 538.8 mA h g−1, even higher than the initial capacity of 409 mA h g−1, which can be attributed to the activation effect. This is in line with the 0.2 C cycling performance. Moreover, taking the reversible capacity at 1 C rate in the initial cycle (400 mA h g−1) and in the 180th cycle (350.3 mA h g−1) into account, the capacity retention is as high as 87.5%, and the Coulombic efficiency remains above 99%, which further indicate that this interesting nanosheet electrode has superior cycling stability even at very high charge–discharge rates.
 | ||
| Fig. 6 (a) Voltage profiles of the electrodes of 2D Ni3Sn2@C@PGC at charge–discharge rates from 0.2 to 5 C. (b) Cycle performance of the 2D Ni3Sn2@C@PGC electrode at current densities of 0.5 C or 1 C. (c) Nyquist plots of 2D Ni3Sn2@C@PGC and 3D Ni3Sn2/C nanoblocks at fresh coin cells over a frequency range from 100 kHz to 0.01 Hz. | ||
To illustrate the reasons for the much higher rate performance of the 2D Ni3Sn2@C@PGC nanosheets than the Ni3Sn2/C nanoblocks, EIS measurements were carried out of the nanosheets and nanoblocks at fresh coin cells. As shown in Fig. 6c, the two impedance spectra have similar features: a medium-to-high-frequency depressed semicircle and a low-frequency linear tail. The high-frequency semicircle is caused by summation of the contact, the SEI resistance Rf, and the charge-transfer impedance Rct on the electrode/electrolyte interface, and the low-frequency linear tail corresponds to the Warburg impedance (Zw) associated with diffusion of lithium ions in the bulk electrode (Re).39,59,60 According to Fig. 6c, the semicircle diameter of the 2D Ni3Sn2@C@PGC electrode is significantly smaller than that of the Ni3Sn2/C nanoblocks, implying that the Ni3Sn2@C@PGC nanosheets have much higher electrical conductivity and much reduced charge-transfer resistances in the electrode.39,59,60
To disclose the mechanism for the excellent electrochemical performance and structure change of the 2D Ni3Sn2@C@PGC nanosheet electrode, the tested cell was disassembled after the cycling test, and the morphology and structure of the nanohybrids were investigated using TEM, as shown in Fig. 7. Although the sample is covered with thin SEI film, we can see clearly that the Ni3Sn2 nanoparticles are still evenly embedded in the 2D carbon nanosheets without any aggregation or pulverization, indicating that the morphology and structure of the nanosheets are well maintained. These results demonstrate that the 2D carbon matrix combined with carbon shells can effectively buffer the volume change of Ni3Sn2 as well as suppress aggregation of Ni3Sn2 during cycling, thus preserving the integrity of the overall electrode.
 | ||
| Fig. 7 TEM and HRTEM images of 2D Ni3Sn2@C@PGC nanosheets after 100 charge–discharge cycles. | ||
As shown, our 2D Ni3Sn2@C@PGC nanosheet electrode displays superior electrochemical performance and structural stability, which can be attributed to the following factors: (1) the active-inactive Ni3Sn2 intermetallic compound. The lithiation of the Sn phase to form Lix<4.4Sn releases the metal component Ni to the surrounding environment, and Ni as a matrix can absorb the massive volume changes occurring during the lithiation/delithiation processes. Uniform distribution of the electrically conducting Ni nanoparticles in close contact with LixSn binaries could contribute to the electrical integration in the anode. Moreover, additional Ni phase could also function as an inactive matrix to inhibit Sn aggregation. All these advantages are beneficial for achievement of outstanding cycling stability. (2) The dual carbon-encapsulated structure. The onion-like carbon shells can effectively prevent Ni3Sn2 nanoparticles from aggregation as well as direct exposure to electrolyte, which is favorable for maintaining the structural and interfacial stabilization of Ni3Sn2 nanoparticles.10,39 Meanwhile, the flexible and conductive graphitic carbon nanosheets can accommodate the mechanical stress caused by volume change of Ni3Sn2 nanoparticles as well as further assist onion-like carbon shells to inhibit aggregation of Ni3Sn2 nanoparticles and thus maintain the structural and electrical integrity of the Ni3Sn2@C@PGC electrode during the charge and discharge processes. (3) The 2D porous and integrative structure. The 2D integrative feature and porous nature of the nanosheets as thin as only about 30 nm provide a large electrode–electrolyte contact area, which can render easy access of liquid electrolyte to the bulk of the electrode material to provide fast transport channels for the lithium ions and offer efficient transport pathways for ion diffusion toward the deep portions of the electrodes. Moreover, it would be easy for lithium ions, electrochemically adsorbed on both sides of 2D ultrathin nanosheets, to have high reversible storage capability. In addition, the well-graphitized micrometer-sized carbon nanosheets can form an effective and continuous conductive network, which gives rise to a very high electrical conductivity of the overall electrode and thus is highly favorable for improving the rate capability of the Ni3Sn2@C@PGC electrode.10,39 Based on the analyses presented above, we believe our material is of enhanced structural stability and integrity, as well as having excellent kinetics for lithium ion and charge transportation, thus its lithium storage property is improved remarkably.
4. Conclusions
In summary, carbon-coated Ni3Sn2 nanoparticles embedded in 2D porous carbon nanosheets (2D Ni3Sn2@C@PGC) with superior electrochemical performance were fabricated via a facile and scalable method, which involves in situ synthesis of 2D Ni@C@PGC and chemical vapor transformation processes. This unique hybrid nanostructure consists of very thin 2D porous graphitic carbon nanosheets with a thickness of less than 30 nm, in which Ni3Sn2 nanoparticles (5–20 nm) coated with thin onion-like carbon shells (∼2 nm) are homogeneously embedded. In this unique dual encapsulation structure, the onion-like carbon shells can effectively prevent Ni3Sn2 nanoparticles from aggregation as well as direct exposure to electrolyte and thus maintain structural and interfacial stabilization of Ni3Sn2 nanoparticles. Meanwhile, the flexible and conductive graphitic carbon nanosheets with high porosity can not only accommodate the mechanical stress caused by volume change of Ni3Sn2 nanoparticles, but also provide large surface area and short diffusion pathway for transfer of lithium ions and electrons, thereby leading to excellent structural and electrical integrity of the overall electrode during the charge–discharge processes. Therefore, this unique structure demonstrates a high reversible capacity up to 585.3 mA h g−1 at a current density of 0.2 C (1 C = 570 mA h g−1) after 100 cycles, a high rate capability (484, 424, 378, 314 and 188 mA h g−1 at 0.2, 0.5, 1, 2 and 5 C, respectively), excellent capacity retention rate (about 90% after a two-period rate test, a total of 320 cycles), and superior cycling performance at a very high rate (350.3 mA h g−1 at a high rate of 1 C after 180 cycles). The strategy we have exhibited here could be extended to construct many other two-dimensional materials and carbon-encapsulated structures for important applications in other scientific fields, such as supercapacitors, catalysts, fuel cells and sensors.Acknowledgements
The authors acknowledge the financial support by the National Natural Science Foundation of China (no. 51422104, no. 51472177 and no. 51272173) and Foundation for the Author of National Excellent Doctoral Dissertation of China (no. 201145), Program for New Century Excellent Talents in University (NCET-12-0408), Natural Science Foundation of Tianjin City (no. 12JCYBJC11700), Elite Scholar Program of Tianjin University, Innovation Foundation of Tianjin University and National Basic Research Program of China (2010CB934700).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- B. Wang, X. Li, T. Qiu, B. Luo, J. Ning, J. Li and L. Zhi, Nano Lett., 2013, 13, 5578 CrossRef CAS PubMed.
- B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang and L. Zhi, ACS Nano, 2013, 7, 1437 CrossRef CAS PubMed.
- D. J. Xue, S. Xin, Y. Yan, K. C. Jiang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 2512 CrossRef CAS PubMed.
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, Chem. Mater., 2012, 24, 3738 CrossRef CAS.
- Y. Yu, L. Gu, C. Zhu, P. A. van Aken and J. Maier, J. Am. Chem. Soc., 2009, 131, 15984 CrossRef CAS PubMed.
- Y. Yu, L. Gu, C. Wang, A. Dhanabalan, P. A. van Aken and J. Maier, Angew. Chem., Int. Ed., 2009, 48, 6485 CrossRef CAS PubMed.
- Y. Yu, L. Gu, X. Lang, C. Zhu, T. Fujita, M. Chen and J. Maier, Adv. Mater., 2011, 23, 2443 CrossRef CAS PubMed.
- J. Qin, C. He, N. Zhao, Z. Wang, C. Shi, E. Z. Liu and J. Li, ACS Nano, 2014, 8, 1728 CrossRef CAS PubMed.
- I. A. Courtney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045 CrossRef CAS PubMed.
- J. Yang, M. Winter and J. O. Besenhard, Solid State Ionics, 1996, 90, 281 CrossRef CAS.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470 CrossRef CAS PubMed.
- R. Hu, M. Zhu, H. Wang, J. Liu and J. Zou, Acta Mater., 2012, 60, 4695 CrossRef CAS PubMed.
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652 CrossRef CAS PubMed.
- Y. Zou and Y. Wang, ACS Nano, 2011, 5, 8108 CrossRef CAS PubMed.
- B. Wang, B. Luo, X. Li and L. Zhi, Mater. Today, 2012, 15, 544 CrossRef CAS.
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160 CrossRef CAS.
- M. Chamas, M. T. Sougrati, C. Reibel and P. E. Lippens, Chem. Mater., 2013, 25, 2410 CrossRef CAS.
- Y. Gu, F. Wu and Y. Wang, Adv. Funct. Mater., 2013, 23, 893 CrossRef CAS.
- P. Chen, L. Guo and Y. Wang, J. Power Sources, 2013, 222, 526 CrossRef CAS PubMed.
- J. Hassoun, S. Panero, P. Simon, P. L. Taberna and B. Scrosati, Adv. Mater., 2007, 19, 1632 CrossRef CAS.
- X. L. Wang, W. Q. Han, J. Chen and J. Graetz, ACS Appl. Mater. Interfaces, 2010, 2, 1548 CAS.
- K. Hirai, T. Ichitsubo, T. Uda, A. Miyazaki, S. Yagi and E. Matsubara, Acta Mater., 2008, 56, 1539 CrossRef CAS PubMed.
- M. E. Stournara, P. R. Guduru and V. B. Shenoy, J. Power Sources, 2012, 208, 165 CrossRef CAS PubMed.
- Z. Lu, J. Zhu, D. Sim, W. Zhou, W. Shi, H. H. Hng and Q. Yan, Chem. Mater., 2011, 23, 5293 CrossRef CAS.
- X. Wang, X. L. Wu, Y. G. Guo, Y. Zhong, X. Cao, Y. Ma and J. Yao, Adv. Funct. Mater., 2010, 20, 1680 CrossRef CAS.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li and I. A. Aksay, ACS Nano, 2010, 4, 1587 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085 CrossRef CAS.
- B. Jang, M. Park, O. B. Chae, S. Park, Y. Kim, S. M. Oh and T. Hyeon, J. Am. Chem. Soc., 2012, 134, 15010 CrossRef CAS PubMed.
- J. Liu and X. W. Liu, Adv. Mater., 2012, 24, 4097 CrossRef CAS PubMed.
- Z. Pan, N. Liu, L. Fu and Z. Liu, J. Am. Chem. Soc., 2011, 133, 17578 CrossRef CAS PubMed.
- S. Yin, Y. Zhang, J. Kong, C. Zou, C. M. Li, X. Lu and X. Chen, ACS Nano, 2011, 5, 3831 CrossRef CAS PubMed.
- J. Y. Lee, D. S. Kim and J. Park, Chem. Mater., 2007, 19, 4663 CrossRef CAS.
- J. Park, H. Zheng, Y. W. Jun and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 13943 CrossRef CAS PubMed.
- M. V. Kovalenko, D. V. Talapin, M. A. Loi, F. Cordella, G. Hesser, M. I. Bodnarchuk and W. Heiss, Angew. Chem., Int. Ed., 2008, 47, 3029 CrossRef CAS PubMed.
- Y. M. Chen, X. Y. Li, X. Y. Zhou, H. M. Yao, H. T. Huang, Y. W. Mai and L. M. Zhou, Energy Environ. Sci., 2014, 7, 2689 CAS.
- Y. M. Chen, X. Y. Li, K. Park, J. Song, J. H. Hong, L. M. Zhou, Y. W. Mai, H. T. Huang and J. B. Goodenough, J. Am. Chem. Soc., 2013, 135, 16280 CrossRef CAS PubMed.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, ACS Nano, 2013, 7, 4459 CrossRef CAS PubMed.
- S. Wu, Z. Wang, C. He, N. Zhao, C. Shi, E. Liu and J. Li, J. Mater. Chem. A, 2013, 1, 11011 CAS.
- L. Chen, Z. Wang, C. He, N. Zhao, C. Shi, E. Liu and J. Li, ACS Appl. Mater. Interfaces, 2013, 5, 9537 CAS.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336 CrossRef CAS.
- J. Hassoun, G. Derrien, S. Panero and B. Scrosati, Adv. Mater., 2008, 20, 3169 CrossRef CAS.
- N. Caiulo, C. H. Yu, K. M. K. Yu, C. C. Lo, W. Oduro, B. Thiebaut and S. C. Tsang, Adv. Funct. Mater., 2007, 17, 1392 CrossRef CAS.
- A. A. El-Gendy, V. O. Khavrus, S. Hampel, A. Leonhardt, B. Buchner and R. Klingeler, J. Phys. Chem. C, 2010, 114, 10745 CAS.
- C. H. Huang, R. A. Doong, D. Gu and D. Zhao, Carbon, 2011, 49, 3055 CrossRef CAS PubMed.
- X. F. Li, A. Dhanabalan, L. Gu and C. L. Wang, Adv. Energy Mater., 2012, 2, 238 CrossRef CAS.
- X. F. Li, Y. Zhong, M. Cai, M. P. Balogh, D. N. Wang, Y. Zhang, R. Y. Li and X. L. Sun, Electrochim. Acta, 2013, 89, 387 CrossRef CAS PubMed.
- Y. H. Xu, J. C. Guo and C. S. Wang, J. Mater. Chem., 2012, 22, 9562 RSC.
- J. Hassoun, G. A. Elia, S. Panero and B. Scrosati, J. Power Sources, 2011, 196, 7767 CrossRef CAS PubMed.
- D. B. Polat, J. Lu, A. Abouimrane, O. Keles and K. Amine, ACS Appl. Mater. Interfaces, 2014, 6, 10877 CAS.
- M. Kotobuki, N. Okada and K. Kanamura, Chem. Commun., 2011, 47, 6144 RSC.
- W. J. Cui, F. Li, H. J. Liu, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2009, 19, 7202 RSC.
- B. Feng, J. Xie, G. S. Cao, T. J. Zhu and X. B. Zhao, New J. Chem., 2013, 37, 474 RSC.
- C. M. Park and K. J. Jeon, Chem. Commun., 2011, 47, 2122 RSC.
- X. L. Wang, H. Chen, J. Bai and W. Q. Han, J. Phys. Chem. Lett., 2012, 3, 1488 CrossRef CAS.
- D. H. Nam, R. H. Kim, C. L. Lee and H. S. Kwon, J. Electrochem. Soc., 2012, 159, A1822 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, X. Ren and G. Yi, J. Electrochem. Soc., 2011, 158, A1404 CrossRef CAS PubMed.
- Y. M. Chen, Z. G. Lu, L. M. Zhou, Y. W. Mai and H. T. Huang, Nanoscale, 2012, 4, 6800 RSC.
- Y. M. Chen, Z. G. Lu, L. M. Zhou, Y. W. Mai and H. T. Huang, Energy Environ. Sci., 2012, 5, 7898 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07520j |
| This journal is © The Royal Society of Chemistry 2014 |