
Anionic waterborne polyurethane dispersion from a bio-based ionic segment
Ruqi Chena,
Chaoqun Zhanga and
Michael R. Kessler *abcd
*abcd
aDept. of Materials Science and Engineering, Iowa State University, Ames, IA, USA
bDept. of Mechanical Engineering, Iowa State University, Ames, IA, USA
cSchool of Mechanical and Materials Engineering, Washington State University, Pullman, WA, USA. E-mail: MichaelR.Kessler@wsu.edu
dAmes Laboratory, US Dept. of Energy, Ames, IA, USA
First published on 4th August 2014
Abstract
Anionic waterborne polyurethane dispersions were prepared from ring-opening epoxidized linseed oil with glycol and hydrochloric acid followed by saponification, step-growth polymerization, and ionomerization. When the intermediate bio-based polyhydroxy fatty acid has an OH functionality of 4.8, the fatty acid can crosslink, and its carboxylic groups are able to provide surface charge for the stabilization of the resulting polymer in the water phase. Two novel anionic waterborne polyurethane dispersions, one with and one without additional castor oil, were successfully prepared and compared to a conventional control sample. Films from the polyurethane dispersions were obtained by casting the dispersions into molds and subsequently characterized by differential scanning calorimetry, dynamic mechanical analysis, ethanol absorption and uptake, thermogravimetric analysis, and tensile stress–strain tests. The castor oil containing polymer displayed a decrease in glass transition temperature, tensile strength, and Young's modulus, but an increase in elongation compared to the control sample. The sample without the castor oil behaved like a brittle, glassy material with higher Young's modulus and lower ductility because of its relatively high crosslinking density. This work proves the viability of incorporating vegetable-oil based polyhydroxy fatty acids as ionic segments into anionic waterborne polyurethane dispersions.
Introduction
Over the past few decades, polyurethanes (PUs) have been employed in various applications, such as coatings, adhesives, sealants, foams, elastomers, and others.1 Environmental concerns regarding conventional, solvent-based PUs have led to the development of waterborne, anionic and cationic polyurethane dispersions (PUDs) because of their environmentally-friendly nature and low content of volatile organic chemicals (VOCs) and hazardous air pollutants (HAPs).2 The basic chemical components of waterborne PUDs are commonly known building blocks, including polyols, isocyanates, catalysts, additives, and others.3 A general awareness of the finite crude oil reserves has triggered extensive studies focused on using bio-based resources for the manufacture of polyols rather than petroleum-based materials. Vegetable oil is one of the most promising options because of its ready availability, relatively low cost, environmental sustainability, and low eco-toxicity. This work has led to the successful development of anionic PUDs from methoxylated soybean oil polyols.4 Castor oil, which contains approx. 2.4 hydroxyl groups per molecule, is one of the vegetable oils suitable to produce polyols for PUD preparation.5The distinct feature of waterborne PUDs is the presence of external or internal emulsifiers that provide stability to the hydrophobic polyurethane dispersed in the water phase. Internal emulsifiers, which are incorporated into the polymer network structure, are favored because the particle sizes in the resulting PUDs are smaller, leading to better stability. For anionic PUDs, dimethylol propionic acid (DMPA) is typically used as an internal emulsifier. DMPA serves as a chain extender in reaction with isocyanates, and its carboxylic acid group in ionic form provides surface charge to PU particles, stabilizing PUDs in the water phase.5 However, DMPA is not derived from bio-renewable resources so that the bio-content of the waterborne PUD system is limited.
Vegetable oils are potential starting materials to replace DMPA because they are triglycerides constituted of glycerol and three fatty acid chains, typically containing unsaturated carbon–carbon double bonds that are available for modifications. Hydroformylation,6,7 epoxidation followed by ring-opening,8–10 ozonolysis,11 and other methods can be employed to convert the double bonds into hydroxyl groups, which react with isocyanates to form urethane bonds. In addition, vegetable oil-based polyols can be saponified into polyhydroxy fatty acids12 which exhibit similar properties as DMPA as long as the average functionality of the hydroxyl exceeds 2.
In this work, epoxidized linseed oil (ELO) was ring-opened by glycol and HCl, followed by saponification into polyhydroxy fatty acid (FA). Oligomerization during the glycol ring-opening step created FAs with a number average molecular weight of 1147 g mol−1. The calculated average functionality of the hydroxyl group was 4.8 providing the FA with sufficient functionality to serve as a crosslinking agent. The presence of a carboxylic acid group also allowed the FA to be employed as an ionic segment. Two novel types of DMPA-free, waterborne PUDs were developed: CasFAD was obtained from castor oil, FA and isophorone diisocyanate (IPDI), while FAD was prepared from FA and IPDI. Both PUDs were stable and had particle sizes of 35.11 nm and 56.11 nm, respectively. The thermal and mechanical properties of the resulting PUD films were characterized and compared to those of CasPAD, which was obtained from castor oil, DMPA and IPDI. Literature research indicates that this is the first report on the substitution of DMPA by an internal emulsifier/ionic segment based on bio-renewable resources.13–19 This work proves the feasibility of using vegetable oil-based polyhydroxy fatty acids to create stable, anionic waterborne PUDs.
Results and discussions
Preparation and properties of FA
The preparation of FA started with the ring-opening of ELO by glycol and HCl, followed by saponification into fatty acid chains. Glycol, which is an effective ring-opening agent,20 inevitably causes oligomerization among oil molecules.2 Consequently, multiple oil molecules can be coupled via ether linkages so that the obtained fatty acids exhibit higher than predicted molecular weights. The GPC data collected (Table 1) confirmed that the number average molecular weight reached 1147 g mol−1, which indicated that on average approximately three fatty acid chains were coupled. Noticeably, FA's polymeric dispersion index (PDI) of 1.8 indicated a broad molecular weight distribution, also indicative of oligomerization. The functionality of OH was estimated by:
| OH number (mg KOH per g) | fn | Acid number (mg KOH per g) | Viscosity (Pa s) at 40 °C | Mn | Mw | PDI |
|---|---|---|---|---|---|---|
| 235.1 | 4.8 | 139.3 | 4.6 | 1147 | 2068 | 1.8 |
FA exhibited an average functionality of 4.8, ensuring the formation of a crosslinking structure in PU. In addition, its high acid number of 139.3 mg KOH per g was a key factor in this work, because the carboxylic group was neutralized into an ionic form, stabilizing the dispersion in the water phase. The presence of hydroxyl groups and carboxylic groups ensured high intermolecular hydrogen bonding, so that the prepared FAs exhibited a viscosity as high as 4.6 Pa s at 40 °C.
Fig. 2 shows FTIR spectra of FA and ELO. For FA, a new broad trough between 3600 cm−1 to 2500 cm−1 emerged, which was attributed to the overlapped signal from –OH stretching of the hydroxyl and carboxylic groups, confirming the presence of the desired functional groups.21 The peak at 823 cm−1, assigned to the epoxy group, disappeared for FA because of the ring-opening reactions.22 Additionally, carbonyl absorption peak at 1740 cm−1 for ELO shifted to 1725 cm−1 for FA, indicating the completion of saponification. These changes in the FTIR spectra confirmed the completion of oxirane ring reduction and saponification.
 | ||
| Fig. 1 Visual appearance of (a) CasPAD, (b) CasFAD, and (c) FAD. | ||
 | ||
| Fig. 2 FTIR spectra of ELO and FA from ELO. | ||
1H-NMR spectra of FA and ELO are shown in Fig. 3. Signal 1 (5.25 ppm) and signal 2 (4.0–4.4 ppm), which are attributed to the backbone glyceride structure, were not prominent for FA.23 This confirmed the cleavage of the triglyceride structure into fatty acid chains during the saponification step. Signal 3 (2.8–3.2 ppm) for ELO was attributed to epoxy groups, and e1 (2.9 ppm) in particular was attributed to the epoxy group anchored to the fatty acid arm with only one derivative group.9 The presence of e1 in FA indicated that a trace amount of the epoxy groups was not reduced. In addition, signal 4 (3.2–4.0 ppm) was mainly created by multiple forms of ethylene protons of glycol units in FA, indicating the complex nature of the ring-opened product. Also, signal 5 (2.3 ppm), assigned to the methylene group adjacent to the carboxyl group –CH2–COO–, did not significantly shift when ELO was converted to FA.24
 | ||
| Fig. 3 FTIR spectra of FA and ELO. | ||
Preparation and properties of PUDs
The TEM images of the different PUDs are shown in Fig. 4, while their size distributions are presented in Fig. 5. Statistical data regarding average particle diameters and polydispersity indices (PDI) were determined using Dispersion Technology Software (Malvern Instruments Ltd.) and are summarized in Table 2. CasPAD and CasFAD exhibited average particle sizes of 29.92 nm and 35.15 nm, respectively. The significant difference was attributed to the fact that the ionic segment FA (Mn = 1147 g mol−1) was substantially larger than DMPA (Mn = 134 g mol−1), based on their molecular weights. However, both exhibited similar PDIs, as both PUD systems started with a pre-polymerization reaction between castor oil (PDI = 1.02) and excess IPDI, which yielded uniform core seeds. Compared to CasFAD, FAD exhibited larger average particle sizes (56.11 nm) and a higher PDI (0.274), resulting from the difference in synthetic route. The preparation of CasFAD started from a castor oil/IPDI pre-polymer with uniform structure, while FAD preparation directly started with a solution polymerization reaction between IPDI and FA with its complicated composition. The particle sizes of the obtained PUDs may also have been affected by other factors such as hydrophilicity, pre-polymer viscosity, ionic group position, and others.4,25,26 However, the PUD particle size had no direct impact on the physical properties of the resulting PU films.4 | ||
| Fig. 4 TEM images of PUD particles of (a) CasPAD, (b) CasFAD and (c) FAD. | ||
 | ||
| Fig. 5 Particle size distribution for PUDs. | ||
| PUD | Z-average size (nm) | Polydispersity index |
|---|---|---|
| CasPAD | 29.92 | 0.232 |
| CasFAD | 35.15 | 0.221 |
| FAD | 56.11 | 0.274 |
Polyurethane properties
Differential scanning calorimetry (DSC) thermograms of the obtained PUD films are shown in Fig. 6. Each PUD exhibited only one Tg and no melting or crystallizing peak, indicating that all PUs had homogenous, amorphous structure. CasPAD had a Tg of 12.9 °C, which matched reported data.5 When DMPA was replaced by FA, the hard segment content dropped from 52.1% to 36.4%. As a result, the chain mobility was less restricted in CasFAD so that it exhibited a lower Tg of 5.94 °C. On the other hand, FAD had a significantly higher Tg (17.62 °C) than CasFAD. Although castor oil and FA have the same number of hydroxyl groups, the OH number of CasFAD was estimated to be 187.9 mg KOH per g, while the OH number of the FAD system was 235.1 mg KOH per g. The substantial difference in OH number led to different crosslinking densities, which directly influenced the respective Tgs.4 In addition, the hard segment content of FAD was higher than that of CasFAD, which also influenced the Tg.27 | ||
| Fig. 6 DSC scans of PUD films. | ||
Fig. 7 depicts the temperature dependence of the storage modulus (E′) and the loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of the PUD films. The storage moduli of all films were similar at temperatures below 0 °C. After a dramatic drop in the value of E′, the materials entered into a rubbery stage from the glassy stage. A rubbery plateau was observed for CasPAD and CasFAD, while FAD yielded at elevated temperatures. The glass transition temperatures (obtained from the maximum of the tanδ curve) and E′ at room temperature are summarized in Table 3. The difference between Tgs obtained from DSC and DMA are caused by the different nature of the two measurements. DSC measures the heat capacity change from frozen to unfrozen chains, while DMA measures the change in mechanical response of polymer chains to heating.28 All PUD films exhibited only one tanδ peak, indicating the homogeneous nature of the PUs. The hard segment content of CasFAD was substantially lower than that of CasPAD (36.4% compared to 52.1%). The higher hard segment content contributed to the stiffness of the network structure, directly influencing both storage modulus and Tg.29 Comparing CasFAD and FAD, the OH number of the starting monomers directly influenced glass transition temperature and storage modulus. Higher OH numbers led to higher crosslinking densities so that less dangling chains acted as plasticizers in the network structure.
δ) of the PUD films. The storage moduli of all films were similar at temperatures below 0 °C. After a dramatic drop in the value of E′, the materials entered into a rubbery stage from the glassy stage. A rubbery plateau was observed for CasPAD and CasFAD, while FAD yielded at elevated temperatures. The glass transition temperatures (obtained from the maximum of the tanδ curve) and E′ at room temperature are summarized in Table 3. The difference between Tgs obtained from DSC and DMA are caused by the different nature of the two measurements. DSC measures the heat capacity change from frozen to unfrozen chains, while DMA measures the change in mechanical response of polymer chains to heating.28 All PUD films exhibited only one tanδ peak, indicating the homogeneous nature of the PUs. The hard segment content of CasFAD was substantially lower than that of CasPAD (36.4% compared to 52.1%). The higher hard segment content contributed to the stiffness of the network structure, directly influencing both storage modulus and Tg.29 Comparing CasFAD and FAD, the OH number of the starting monomers directly influenced glass transition temperature and storage modulus. Higher OH numbers led to higher crosslinking densities so that less dangling chains acted as plasticizers in the network structure.
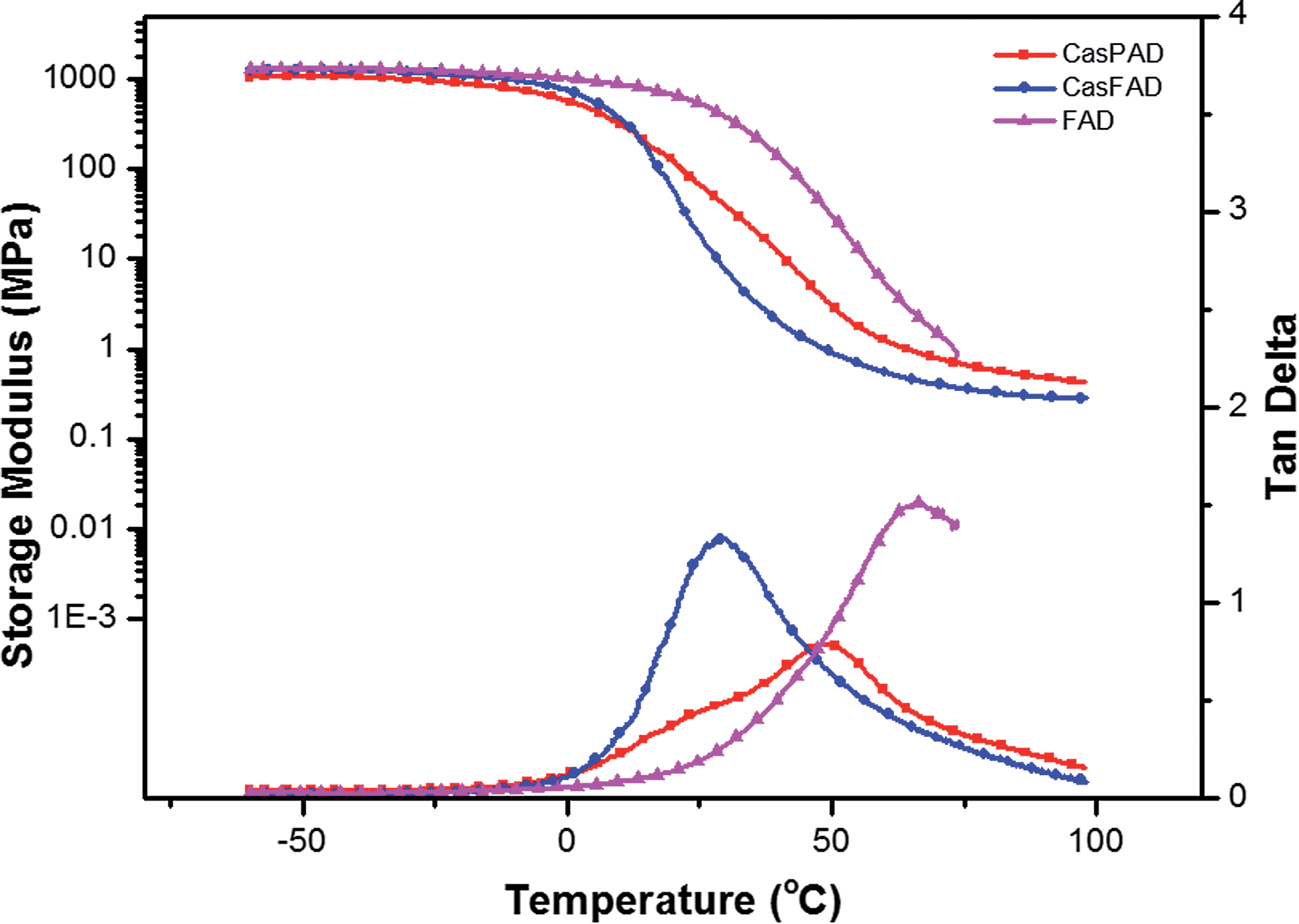 | ||
| Fig. 7 Storage modulus and loss factor of PU films as functions of temperature. | ||
| Hard segment content (wt%) | Tg (°C) DSC | Tg (°C) DMA | E′ at room temperature (MPa) | T10 (°C) | T50 (°C) | |
|---|---|---|---|---|---|---|
| CasPAD | 52.1 | 12.9 | 49.1 | 68.3 | 305.2 | 383.5 |
| CasFAD | 36.4 | 5.9 | 29.4 | 19.0 | 302.5 | 379.1 |
| FAD | 49.3 | 17.6 | 65.7 | 539.3 | 301.32 | 376.6 |
The thermal degradation profiles for the obtained PUDs are plotted in Fig. 8. In general, PUs exhibit three stages of thermal decomposition in air atmosphere.30 The first weight loss domain was observed between 200 and 300 °C and assigned to the dissociation of the labile urethane bonds.31 The dissociation of urethanes to isocyanates and alcohols, the formation of primary amines and olefins, and the formation of secondary amines were reported as three possible mechanisms by Wang et al.20 The weight percentage of IPDI in CasPAD and CasFAD, which determined the amount of urethane bonds, differed remarkably: CasPAD contained 27.3% IPDI, while CasFAD contained 23.7% IPDI. Consequently, CasFAD exhibited lower weight loss at 300 °C than CasPAD, and FAD underwent greater weight loss during the urethane bond decomposition stage than CasFAD. The second thermal degradation stage between 300 °C and 450 °C resulted from chain scission of the oil structure. The last stage, above 450 °C, was attributed to further thermo-oxidation of the PUs in air. The temperatures of 10% and 50% weight loss (T10 and T50) are summarized in Table 3. CasFAD exhibited better thermal resistance than CasPAD because of its lower content of labile urethane bonds.30 FAD showed better thermal resistance after urethane bond dissociation than CasFAD, attributed to higher crosslinking densities caused by higher OH numbers of the polyol component.5
 | ||
| Fig. 8 TGA curves of PU films. | ||
Fig. 9 shows tensile stress–strain curves for all PUD films. Young's moduli, tensile strength at break, elongation at break, and toughness are summarized in Table 4. CasFAD exhibited lower tensile strength, lower Young's modulus, but higher ductility than CasPAD, which met expectation because of the lower hard segment content in CasFAD. CasFAD and CasPAD did not differ significantly in toughness (14.31 MPa and 11.76 MPa, respectively), while the toughness of FAD was substantially lower. Because FAD had a relatively higher content of carboxylic acid, the mechanical properties may have been compromised.32 In general, brittle materials exhibit low toughness, while ductile materials are tough.5
 | ||
| Fig. 9 Stress–strain curves for PU films. | ||
| E (MPa) | σb (MPa) | εb (%) | Toughness (MPa) | |
|---|---|---|---|---|
| CasPAD | 13.6 | 5.5 | 381.7 | 14.31 |
| CasFAD | 2.3 | 3.5 | 642.9 | 11.76 |
| FAD | 77.4 | 6.8 | 19.3 | 0.83 |
Conclusions
In this work, ELO was ring-opened by glycol and HCl, followed by saponification to yield polyhydroxy fatty acid (FA). FA was successfully utilized to replace DMPA in castor oil-based anionic polyurethane dispersion system. The obtained CasFAD had an average particle size diameter of 35.15 nm, while the control sample CasPAD had an average particle size of 29.92 nm. It was shown that FA can serve as both polyol component and as ionic segment in the preparation of PUDs. The resulting FAD had an average particle size of 56.11 nm. CasFAD exhibited lower tensile strength and Young's modulus than CasPAD because of its lower hard segment content. Nonetheless, CasFAD and CasPAD were comparable in terms of toughness. The mechanical properties of FAD were compared with those of CasFAD. FAD was relatively brittle and less tough because of its high OH number and acid content. In general, vegetable oil-based polyhydroxy fatty acids were successfully incorporated into anionic PU systems as ionic segments.Materials and methods
Materials
Epoxidized linseed oil (ELO) ( , functionality 5.5, oxygen content 9.0–9.5%) was kindly provided by American Chemical Service Inc., Griffith, IN. Magnesium sulfate (MgSO4), potassium hydroxide (KOH), ethylene glycol, hydrochloric acid (HCl), and methyl ethyl ketone (MEK) were purchased from Fisher Scientific Company (Fair Lawn, NJ). Tetrafluoroboric acid solution (48% in H2O), 2,2-bis(hydroxymethyl)propionic acid (DMPA), isophorone diisocyanate (IPDI), dibutyltin dilaurate (DBTDL), sodium bicarbonate (NaHCO3), triethylamine (TEA), and castor oil were obtained from Sigma-Aldrich (Milwaukee, WI). Ethanol was purchased from Decon Laboratories Inc., King of Prussia, PA. All materials were used as received without further purification.
, functionality 5.5, oxygen content 9.0–9.5%) was kindly provided by American Chemical Service Inc., Griffith, IN. Magnesium sulfate (MgSO4), potassium hydroxide (KOH), ethylene glycol, hydrochloric acid (HCl), and methyl ethyl ketone (MEK) were purchased from Fisher Scientific Company (Fair Lawn, NJ). Tetrafluoroboric acid solution (48% in H2O), 2,2-bis(hydroxymethyl)propionic acid (DMPA), isophorone diisocyanate (IPDI), dibutyltin dilaurate (DBTDL), sodium bicarbonate (NaHCO3), triethylamine (TEA), and castor oil were obtained from Sigma-Aldrich (Milwaukee, WI). Ethanol was purchased from Decon Laboratories Inc., King of Prussia, PA. All materials were used as received without further purification.
Preparation of highly branched fatty acid
The preparation of highly branched fatty acids followed three steps:(1) Ring-opening of ELO with glycol: 100 g of epoxidized linseed oil (0.080 mol,  of ELO was 1255 g mol−1 according to GPC data) was mixed with 200 g of glycol (3.2 mol) in a 500 ml round-bottom flask (molar ratio of epoxy:OH was approximately 1:11). After the addition of 0.1 wt% HBF4, the mixture was allowed to react at 98 °C for 2 h under constant stirring; the product was extracted using acetone, then washed with saturated NaCl solution, and the organic layer was used in the next step.
of ELO was 1255 g mol−1 according to GPC data) was mixed with 200 g of glycol (3.2 mol) in a 500 ml round-bottom flask (molar ratio of epoxy:OH was approximately 1:11). After the addition of 0.1 wt% HBF4, the mixture was allowed to react at 98 °C for 2 h under constant stirring; the product was extracted using acetone, then washed with saturated NaCl solution, and the organic layer was used in the next step.
(2) Ring-opening of unreacted epoxide ring with HCl: 50 g of HCl was added to the acetone solution obtained in previous step and reacted at 40 °C for 1 h. Ethyl acetate was used to extract the polyol. Sodium bicarbonate and sodium chloride solution were used sequentially to wash the product until it was neutral. The organic layer was dried over MgSO4 and the solvent was removed subsequently. The effectiveness of the additional ring-opening reaction by HCl was reflected in the increase in OH numbers from 221.9 mg KOH per g to 229.2 mg KOH per g.
(3) Saponification of the polyol: 100 g of polyol was dissolved in ethanol and 50 g of potassium hydroxide was added to trigger saponification at 73 °C. The reaction proceeded for 4 h followed by the addition of HCl to neutralize the reactants until phase separation was observed. The fatty acid was extracted by ethyl acetate and washed three times with saturated NaCl solution. After drying over MgSO4, polyhydroxy fatty acid (FA) with dark brown color was obtained after removal of organic solvent by roto-evaporation. OH number titration, acid number titration, and rheological and GPC tests were performed to characterize the physical properties of the obtained FA and the results are summarized in Table 1. The preparation route for FA is illustrated in Scheme 1.
 | ||
| Scheme 1 Preparation of FA. | ||
Preparation of anionic waterborne polyurethane dispersions (PUDs)
Unsuccessful approaches to prepare PUDs from FA may provide insight into the nature of the PUD synthesis. Xia5 reported in his work that castor oil, DMPA, IPDI were first subjected to polymerization without solvent for 1 h. In our original approach, DMPA was replaced by FA to synthesize CasFAD. The complex composition and high reactivity of FA caused rapid gelation of the mixture within less than 30 s. In order to solve the gelation problem, polymerization was initiated in solvent solution in order to effectively lower the reaction rate. However, the obtained PUD precipitated after removal of the organic solvent, which may have been caused by the significant difference in reactivity between castor oil and FA. Castor oil consists of triglycerides composed of roughly 85% ricinoleic acid with a secondary hydroxyl group at the C12 site, while FA contains primary hydroxyl groups derived from ELO ring-opened by glycol. The fact that the primary hydroxyl group exhibited relatively higher reactivity towards isocyanate than the secondary hydroxyl group resulted in a faster incorporation of FA into the cross-linked network.33 Consequently, the core of the PUD particles was more likely composed of FA, while the shell was mainly made of castor oil. Because FA was the component bearing carboxylic acids, which are critical for the stabilization of the PUD particles in the water phase, insufficient FA content in the outer shell led to weak interactions of the PUD particles with the water molecules, creating an unstable waterborne PUD system. In addition, the fact that the simultaneous polymerization of castor oil, DMPA and IPDI resulted in stable dispersions is not in conflict with this explanation, because DMPA is insoluble in castor oil and IPDI. Therefore, DMPA went through a retarded heterogeneous polymerization, possibly resulting in the higher DMPA content in the outer shell of the PUD particles.A modified synthesis route was therefore chosen and three different types of PUDs were synthesized and determined to be stable after no precipitate was detected within one week of sitting still.
(1) Castor oil – fatty acid PUD (CasFAD): castor oil (5.05 g) and IPDI (3.12 g) were introduced into a two-neck flask equipped with a mechanical stirrer. The flask was placed in an oil bath set at 78 °C and the mixture was stirred for 5 min to ensure homogeneity. Then, 2 drops of DBTDL were added to trigger polymerization. After 1 h, FA (3.30 g) in 25 ml MEK was poured into the pre-polymer and allowed to polymerize for another 4 h. Once the reaction was terminated the reactants were cooled to room temperature and TEA (2 equiv. per mole of COOH from FA) was added under vigorous stirring for 30 min. Finally, 150 ml water was used for the phase reversal and the organic solvent was removed under reduced pressure. The molar ratio of OH of castor oil to OH of FA to NCO of IPDI was 1:1:2. The synthesis route of CasFAD is shown in Scheme 2.
 | ||
| Scheme 2 Preparation of CasFAD. | ||
(2) Fatty acid PUD (FAD): the preparation process started with the solution polymerization of FA (8.71 g), IPDI (4.05 g) and DBTDL in 25 ml MEK for 4 h. The mixture gelled rapidly without solvent, which was attributed to the high reactivity and complicated composition of the FA. The subsequent procedural steps were identical to those taken in the preparation of CasFAD.
(3) Castor oil – DMPA PUD (CasPAD): the preparation method was similar to the one used to prepare CasFAD; however, DMPA was used instead of FA. It is worth mentioning that DMPA is not soluble in MEK so that DMPA was added in its powder form along with MEK.
Fig. 1 shows the visual appearance of the obtained PUDs. With the increasing FA content, which had a dark brown color, the resulting PUDs turned darker. Polyurethane films were obtained by casting the PUD into a Teflon mold and allowing it to dry out at ambient temperature. Heat-drying was performed in a conventional oven at 80 °C overnight. The obtained PU films were cut into desired dimensions for specific characterizations.
Characterization
1H NMR spectra were recorded on a Varian spectrometer (Palo Alto, CA) at 300 MHz for FA and ELO in chloroform-d. A Bruker IFS66V FT-IR spectrometer was employed to record infrared spectra of FA and ELO. The scanning resolution was 4 cm−1 and the scanning range covered 4000 cm−1 to 400 cm−1. The hydroxyl and acid numbers of the fatty acids and polyols were determined using Unilever method and AOCS Official Method Te 1a-64, respectively. The average molecular weight was measured by a Thermo Scientific Dionex Ultimate 3000 GPC (Sunnyvale, CA) equipped with a Shodex Refractive Index (RI). The standard curve was calibrated by polystyrene standard kit. The eluent solvent used was tetrahydrofuran with two Agilent PLgel 3 μm 100 Å 300 × 7.5 mm (p/n PL1110-6320) and one mesopore 300 × 7.5 mm (p/n PL1113-6325). The flow rate of THF was 1.0 ml min−1 and the measurement was conducted at room temperature. The rheological test was performed at 40 °C using an AR2000 (TA Instrument). The rheological behavior of FA was investigated by varying the shear rate from 10 s−1 to 1000 s−1. A JEOL scanning and transmission electron microscope (Japan Electron Optic Laboratories, Peabody, MA) was used to observe the morphology of the PUD particles. Diluted PUD (2 μl with approx. 0.1 wt% solid content) was placed on 200-mesh carbon film grid. After drying, the polymer thin layer was stained by RuO4 before observation. The size distribution of the PUDs was determined by NanoZS90 Zeta-sizer (Worcestershire, UK) at room temperature.Dynamic mechanical analysis (DMA) of the polyurethane films was carried out on a TA Instruments DMA Q800 dynamic mechanical analyzer using a film-tension mode of 1 Hz. Rectangular samples with a length of 15 mm and a width of 10 mm were used for the analysis. The samples were cooled and held isothermally for 3 min at −60 °C before raising temperature to 100 °C at a rate of 3 °C min−1. The glass transition temperatures (Tgs) of the samples were determined by the peaks of the loss modulus curve.
Thermogravimetric analysis (TGA) of the films was conducted on a TA Instrument Q50 (New Castle, DE). The samples were heated from room temperature to 650 °C at a rate of 20 °C min−1 in air with a flow rate of 60 ml min−1. The remaining weight was recorded as a function of temperature and plotted (see Fig. 8).
Differential scanning calorimetry (DSC) was performed on a TA Instrument Q20. The samples were weighed and placed in Tzero Hermetic pans and subsequently heated to 80 °C to eliminate their heat history. The samples were then cooled to −80 °C by manually adding liquid nitrogen to the chamber and re-heated to 80 °C at a programmed rate of 10 °C min−1. The second heating ramp was recorded and glass transition temperature was determined by the midpoint inflection method.
The tensile properties of the PU films were determined with an Instron universal testing machine (model 4502) with a crosshead speed of 100 mm min−1. Rectangular samples with the dimensions 50 mm × 10 mm (length × width) were prepared. At least three replicates were tested for each PU sample. The stress–strain curve was plotted and toughness, representing the ability of a material to absorb energy before fracture, was calculated as the integrated area beneath the curve.
References
- L. Shen, J. Haufe and M. K. Patel, Biofuels, Bioprod. Biorefin., 2010, 4(1), 25–40 CrossRef CAS PubMed.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- K. L. Noble, Prog. Org. Coat., 1997, 32, 131–136 CrossRef CAS.
- Y. S. Lu and R. C. Larock, Biomacromolecules, 2008, 9, 3332–3340 CrossRef CAS PubMed.
- Y. Xia and R. C. Larock, Macromol. Mater. Eng., 2011, 296, 703–709 CrossRef CAS PubMed.
- A. Guo, D. Demydov, W. Zhang and Z. S. Petrovic, J. Polym. Environ., 2002, 10, 49–52 CrossRef CAS.
- P. Kandanarachchi, A. Guo and Z. Petrovic, J. Mol. Catal. A: Chem., 2002, 184, 65–71 CrossRef CAS.
- A. Guo, Y. J. Cho and Z. S. Petrovic, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3900–3910 CrossRef CAS.
- S. D. Miao, S. P. Zhang, Z. G. Su and P. Wang, J. Appl. Polym. Sci., 2013, 127, 1929–1936 CrossRef CAS PubMed.
- M. M. A. Nikje, F. Abedinifar and A. Idris, Arch. Appl. Sci. Res., 2011, 3(3), 383–388 CAS.
- Z. S. Petrovic, W. Zhang and I. Javni, Biomacromolecules, 2005, 6, 713–719 CrossRef CAS PubMed.
- C. Q. Zhang, Y. Xia, R. Q. Chen, S. Huh, P. A. Johnston and M. R. Kessler, Green Chem., 2013, 15, 1477–1484 RSC.
- Y. U. Ahn, S. K. Lee, S. K. Lee, H. M. Jeong and B. K. Kim, Prog. Org. Coat., 2007, 60, 17–23 CrossRef PubMed.
- C. Y. Bai, X. Y. Zhang and J. B. Dai, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 1203–1208 CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS PubMed.
- P. C. Chen, S. C. Wang, J. Z. Hwang, J. T. Yeh, C. Y. Huang and K. N. Chen, J. Chin. Chem. Soc., 2010, 57, 901–908 CAS.
- Y. S. Lu and R. C. Larock, J. Appl. Polym. Sci., 2011, 119, 3305–3314 CrossRef CAS PubMed.
- S. A. Madbouly, J. U. Otaigbe, A. K. Nanda and D. A. Wicks, Macromolecules, 2007, 40, 4982–4991 CrossRef CAS.
- Y. Xia and R. C. Larock, ChemSusChem, 2011, 4, 386–391 CrossRef CAS PubMed.
- C. S. Wang, L. T. Yang, B. L. Ni and G. Shi, J. Appl. Polym. Sci., 2009, 114, 125–131 CrossRef CAS PubMed.
- Y. Marechal, J. Chem. Phys., 1987, 87, 6344–6353 CrossRef CAS PubMed.
- A. Adhvaryu and S. Z. Erhan, Ind. Crops Prod., 2002, 15, 247–254 CrossRef CAS.
- A. Biswas, A. Adhvaryu, S. H. Gordon, S. Z. Erhan and J. L. Willett, J. Agric. Food Chem., 2005, 53, 9485–9490 CrossRef CAS PubMed.
- E. Sharmin, S. M. Ashraf and S. Ahmad, Int. J. Biol. Macromol., 2007, 40, 407–422 CrossRef CAS PubMed.
- S. H. Park, I. D. Chung, A. Hartwig and B. K. Kim, Colloids Surf., A, 2007, 305, 126–131 CrossRef CAS PubMed.
- J. Y. Jang, Y. K. Jhon, I. W. Cheong and J. H. Kim, Colloids Surf., A, 2002, 196, 135–143 CrossRef CAS.
- T. W. Pechar, G. L. Wilkes, B. Zhou and N. Luo, J. Appl. Polym. Sci., 2007, 106, 2350–2362 CrossRef CAS PubMed.
- Y. S. Lu and R. C. Larock, Biomacromolecules, 2007, 8, 3108–3114 CrossRef CAS PubMed.
- A. Zlatanic, Z. S. Petrovic and K. Dusek, Biomacromolecules, 2002, 3, 1048–1056 CrossRef CAS PubMed.
- K. P. Wang and L. T. Yang, Advanced Materials Research, 2011, 197–198, 1196–1200 CAS.
- G. Trovati, E. Ap Sanches, S. C. Neto, Y. P. Mascarenhas and G. O. Chierice, J. Appl. Polym. Sci., 2010, 115, 263–268 CrossRef CAS PubMed.
- L. Gindin, R. Konitsney, J. McLafferty and R. Roesler, US Pat. 20050288431 A1, 2005.
- Y. K. Jhon, I. W. Cheong and J. H. Kim, Colloids Surf., A, 2001, 179, 71–78 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |