
Cross-linked poly(dimethylaminoethyl acrylamide) coated magnetic nanoparticles: a high loaded, retrievable, and stable basic catalyst for the synthesis of benzopyranes in water†
Abstract
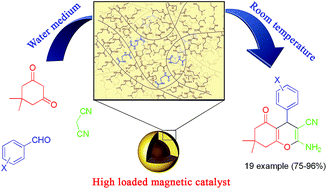
A novel heterogeneous catalyst has been synthesized based on the distillation–precipitation–polymerization of methyl acrylate onto modified magnetic nanoparticles followed by the amidation of the methyl ester groups using N,N-dimethylethylenediamine. The resulting poly(dimethylaminoethyl acrylamide) coated magnetic nanoparticles (MNP@PDMA) catalyst was characterized using an array of sophisticated analytical techniques, including FT-IR, TGA, SEM, TEM, CHN, vibrating sample magnetometer (VSM), and XRD analysis. The resulting heterogeneous base catalyst allowed the performance of a domino Knoevenagel condensation/Michael addition/cycloaddition reaction toward the synthesis of 4H-benzo[b]pyranes in excellent yields using water as the reaction medium. The multilayered and polymeric identity of the coated material on the surface of the magnetic nanoparticles provides many catalytic units resulting in the high loading level and high stability of the catalyst. Straightforward magnetic separation and recycling of the catalyst for up to 5 runs is possible without any significant loss of efficiency.
 Please wait while we load your content...
Please wait while we load your content...

