
Transition metal (Ni, Fe, and Cu) hydroxides enhanced α-Fe2O3 photoanode-based photofuel cell†
Ruifeng Chong‡
,
Zhiliang Wang‡,
Jun Li,
Hongxian Han,
Jingying Shi and
Can Li*
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, 457 Zhongshan Road, Dalian, 116023, China. E-mail: canli@dicp.ac.cn; Web: http://www.canli.dicp.ac.cn Fax: +86-411-84694447; Tel: +86-411-84379070
First published on 8th September 2014
Abstract
Photofuel cells have been demonstrated to be a promising strategy for generating electricity using biomass. Here, we present a photofuel cell with a visible light α-Fe2O3 based photoanode that can be directly powered by a variety of biomasses such as methanol, glycerol, glucose, cellubiose and starch. The photocurrent density and power density of the photofuel cell are significantly enhanced by loading cocatalysts (metal hydroxides, e.g. Ni(OH)2) on the α-Fe2O3 photoanode. The power density of the photofuel cell powered by glucose is enhanced over two times from 0.082 mA cm−2 for α-Fe2O3 to 0.18 mW cm−2 for Ni(OH)2/α-Fe2O3 photoanode.
Introduction
The utilization of abundant biomass as an energy source can prevent environmental pollution and reduce the dependence on fossil resources.1,2 The technological challenge is to sustainably capture biomass energy using green processes. Direct alcohol fuel cells (DAFCs) with potentials of high power density and being pollution-free offer a possible solution to this problem.3,4 Although DAFCs powered by low molecular weight alcohols (usually methanol or ethanol) have been demonstrated to be practically feasible, biomass-derived compounds, such as glucose, polysaccharide, and cellulose, have not been reported as fuels for DAFCs. This is because of the low activity of catalysts in the activation of the C–C bonds and the oxidation of biomass at low temperatures.5,6 Biofuel cells can be powered by various biomasses at low temperatures; however, low electric power output, limited lifetime and rigorous reaction conditions seriously hinder their applications.7,8A photofuel cell (PFC) mainly consists of a semiconductor photoanode, a cathode, electrolyte and fuels.9–14 With PFC technology, biomass as a fuel can be oxidized on the photoanode under light irradiation and oxygen is reduced on the counter electrode through an external circuit, generating electricity. In essence, PFC is different from the fuel cells reported previously and widens the range of biomass fuels for electricity generation. Thus, various biomasses, such as glycerol, glucose, saccharides, proteins, and ammonia, can be used as fuels for PFCs with a TiO2 photoanode. Previous work has demonstrated that PFCs can convert biomass into electricity even at low ambient temperatures. However, TiO2 and WO3 with band gaps of 3.2 eV and 2.8 eV, respectively, use only a small proportion of solar irradiation.15 Therefore, semiconductors with wide ranges of light absorption are highly desired for PFCs.
Hematite (α-Fe2O3), with an optical band gap of 2.1 eV, is a promising material for photoanodes due to its abundance, ecofriendly nature, and photochemical stability in basic electrolytes.16–18 However, its short excited state lifetime (∼1 ps) and the small hole diffusion length (∼2 to 4 nm) significantly hinders its efficiency in charge separation and collection. To overcome these problems, various strategies have been adopted, including reducing the size,19,20 doping21–23 and loading cocatalysts.24–26 Among these strategies, loading cocatalysts is the most efficient method for photoelectrochemical water splitting. However, α-Fe2O3 loaded with an appropriate cocatalyst that can distinctly enhance biomass oxidation has not been reported.
In this work, we present a PFC with α-Fe2O3 based photoanodes that directly converts biomass-derived compounds such as methanol, glucose, glycerol, cellubiose, and starch. To improve the PFC efficiency and the stability of α-Fe2O3 photoanode, transition metal hydroxides Ni(OH)2, Fe(OH)3, and Cu(OH)2 are loaded as biomass oxidation cocatalysts. It was found that cocatalysts can remarkably enhance the photoresponse and stability of bare α-Fe2O3 for biomass oxidation, which demonstrates an example of stimulating a PFC by loading an efficient cocatalyst on the photoanode.
Experimental
Materials
All the chemicals were of analytical grade and were used as purchased. Solutions were prepared using high purity water (Millipore Milli-Q purification system, resistivity > 18 MΩ cm). FTO (fluorine-doped tin oxide) conductive glass was purchased from Nippon Sheet Glass Company 5 (Japan) and was ultrasonically cleaned with acetone, ethanol and deionized water for 20 min each in sequence prior to use.Preparation of the Fe2O3 films
α-Fe2O3 films were deposited on an F-doped SnO2 (FTO) substrate electrode using a modified chemical bath deposition method reported elsewhere.27 Specifically, 0.2 mol L−1 FeCl3 (FeCl3·6H2O, ≥99%, Shanghai Chemical) aqueous solution (10 mL) and 0.3 mol L−1 NH2CONH2 (98%, Shanghai Chemical) were mixed in a 50 mL glass beaker and heated at 100 °C for 4 h. FTO was placed vertically in this beaker with the conducting edge facing the wall of the beaker. After the reaction, the film formed on FTO was thoroughly rinsed with high purity water and annealed at 500 °C for 3 h to obtain the desired phase. Finally, the prepared sample was further annealed at 750 °C for 10 min.Fabrication of M(OH)x/Fe2O3 photoanode
Ni(OH)2 was deposited onto α-Fe2O3 by a successive ionic layer adsorption and reaction method. In a typical synthesis, α-Fe2O3 electrodes were dipped into 0.1 mol L−1 Ni(NO3)2 (≥98.5%, Shanghai Chemical) solution for 40 s, followed by drying with compressed air. Then, the electrodes were dipped into 1 mol L−1 KOH solution for another 40 s and dried with compressed air. Next, the prepared sample was washed with ethanol and dried at 60 °C for 1 h in air. Fe(OH)3/α-Fe2O3 and Cu(OH)2/α-Fe2O3 samples were prepared using the same method without further heat treatment.Characterization of the electrodes
The prepared samples were characterized by X-ray powder diffraction (XRD) on a Rigaku D/Max-2500/PC powder diffractometer using Cu Kα radiation (operating voltage: 40 kV, operating current: 20 mA, scan rate: 5° min−1). The UV-visible diffuse reflectance spectra were recorded on a UV-visible spectrophotometer (JASCO V-550) and calibrated by the Kubelka-Munk method. The morphologies of the electrodes were examined using a Quanta 200 FEG scanning electron microscope (SEM) equipped with an energy dispersive spectrometer (accelerating voltage of 20 kV). Transmission electron microscopy (TEM) images were taken on a Tecnai G2 Spirit (FEI company) using an accelerating voltage of 120 kV. High-resolution transmission electron microscopy (HRTEM) images were acquired on Tecnai G2 F30 S-Twin (FEI company) with an accelerating voltage of 300 kV. The liquid products quantitative analysis was carried out using HPLC (Agilent 1200) with refractive index (RI) detector and ultraviolet (UV, λ = 210 nm) detector for arabinose, erythrose, glyceraldehyde, glycolaldehyde, glycolate and formate. The reactants and products were separated through an ion exclusion column (Alltech OA-1000) heated at 55 °C. The eluent was a solution of H2SO4 (0.005 mol L−1). Products were identified through comparison with standard samples, which were obtained from Sigma-Aldrich.Photoelectrochemical and electrochemical measurements
All the photoelectrochemical measurements were carried out in a three-electrode cell with a flat quartz window to facilitate the illumination of the photoelectrode surface. The working electrode was α-Fe2O3, and Hg/HgCl2 (saturated KCl) and a Pt plate (2 cm × 4 cm) were used as a reference and counter electrode, respectively. A Nafion membrane was used to prevent the crossover between the anode and cathode. The illumination source was a 300 W Xe arc lamp and the light intensity at the surface of the electrodes was 300 mW cm−2. The electrochemical measurements were performed on a CHI 760D electrochemical workstation (CHI, Shanghai) at room temperature. The electrolyte was 1 mol L−1 KOH solution with/without 0.025 mol L−1 glucose or methanol (10 vol%), glycerol (0.025 mol L−1), cellubiose (0.025 mol L−1) and starch (1 wt%).A photofuel cell was constructed using a two-compartment quartz cell with a Nafion membrane as the separator. α-Fe2O3 and Ni(OH)2/α-Fe2O3 photoanodes were used as anodes and a Pt wire was used as the cathode, while a Nafion membrane served as the separator. The distance between the anode and the cathode was 10 cm. The photovoltaic performance of the cells was measured with a Keithley 2400 source measure unit irradiated by AM 1.5 (100 mW cm−2).
The energy efficiency (PEC) and fill factor (FF) of such a device can be estimated by eqn (1) and (2), respectively.
| PEC = POut/ Phν × 100% | (1) |
| FF = (Vmax × Imax)/(Voc × Isc) | (2) |
Results and discussions
The XRD patterns of Fe2O3 film are shown in Fig. 1a. The sample shows diffraction peaks with 2θ at 35.6°and 64.0°, which correspond to the indices of the (110) and (300) planes (PDF no. 840306) of hematite phase, respectively. The UV-visible spectrum (Fig. 1b) gives the bands of α-Fe2O3 at 410 nm and around 600 nm, which corresponds to the direct transition of O2− 2p → Fe3+ 3d and the transition of the spin-forbidden Fe3+ 3d → 3d, respectively.28 Fig. 1c and d illustrate the top-view analysis and morphology of α-Fe2O3 film characterized by SEM. The film consists of α-Fe2O3 nanorod particles with a diameter of 20–30 nm and a length of 60–100 nm. The cross analysis of the film shows a thickness of about 300 nm with a uniform and continuous morphology.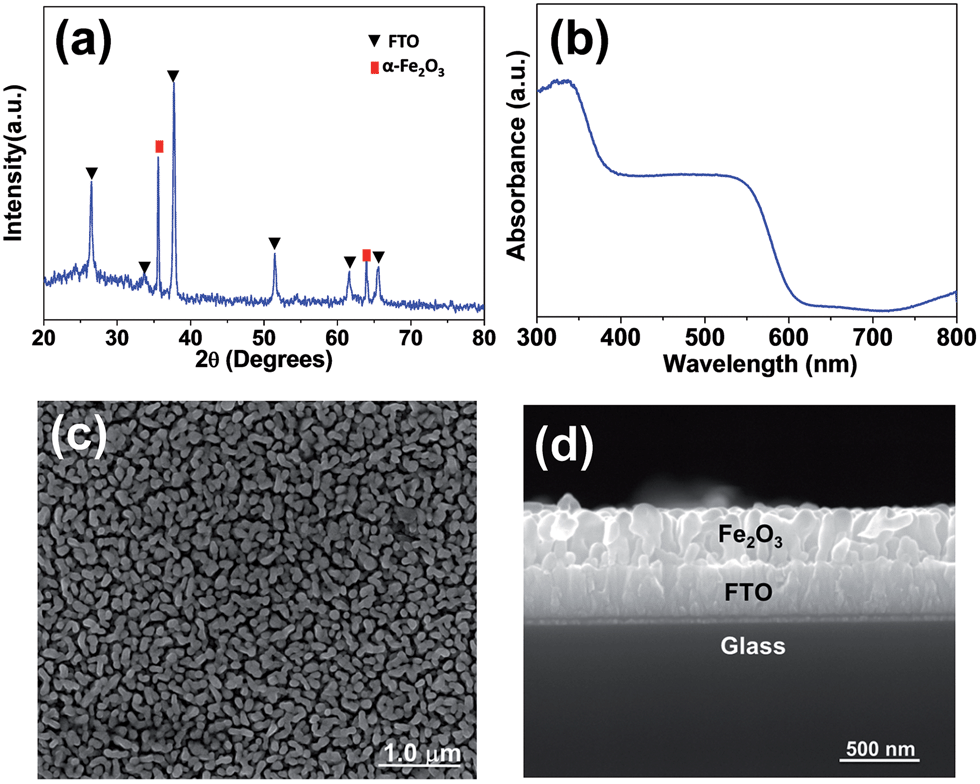 | ||
| Fig. 1 (a) XRD pattern and (b) UV-vis absorption spectra of Fe2O3 film. (c) Top view and (d) side view SEM images of Fe2O3 film. | ||
Fig. 2 shows the dark and photocurrent densities of an α-Fe2O3 photoanode under light illumination. The dark response is negligible up to 0.6 V vs. SCE for both 1 mol L−1 KOH electrolyte and 1 mol L−1 KOH containing glucose. Above 0.6 V vs. SCE, an anodic current is formed due to water or glucose oxidation. This suggested that the α-Fe2O3 photoanode has equal oxidation ability for glucose and water under these conditions. In KOH, the onset potential is shifted to −0.43 V vs. SCE, and the photocurrent is due to water oxidation. When glucose is added to the aqueous KOH electrolyte, the photo-onset potential shifts to a lower value of −0.6 V vs. SCE and the photocurrent density obviously increases. These results indicate that glucose is preferentially oxidized over water and more photo-generated electrons and holes are used for O2 reduction and glucose oxidation. These results also suggest that the α-Fe2O3 photoanode can efficiently oxidize biomass in a photochemical process, which implies that sunlight utilization of PFC could be extended to the visible light region.
 | ||
| Fig. 2 Dark and photocurrent densities for α-Fe2O3 photoanode in 1 mol L−1 KOH electrolyte and in 1 mol L−1 KOH electrolyte containing 0.025 mol L−1 glucose; light source: 300 W Xe lamp; scanning rate: 20 mV s−1. | ||
To improve the activity and stability of the α-Fe2O3 photoanode, transition metal hydroxides Ni(OH)2, Fe(OH)3, and Cu(OH)2 were deposited on α-Fe2O3 film by a successive ionic layer adsorption and reaction method. SEM elemental mapping was carried out to detect the state of the Ni(OH)2 modification on Fe2O3. As shown in Fig. S1,† Ni is distributed randomly on Fe2O3 and the mass ratio of Ni to Fe is ca. 0.24, as detected by EDS. To further study the microstructure of Ni(OH)2/α-Fe2O3, HRTEM analyses were conducted. HRTEM images of a representative Ni(OH)2/α-Fe2O3 photoanode are shown in Fig. 3a and b. The HRTEM image confirms the presence of Ni species on the edge of α-Fe2O3. The magnified HRTEM image in Fig. 3b exhibits fringes with a lattice spacing of ca. 0.37 nm, which corresponds to the (012) plane of α-Fe2O3, while the lattice fringes with a spacing of 0.218 and 0.154 nm are consistent with the (103) and (300) planes of the 3Ni(OH)2·2H2O (α-Ni(OH)2). The EDS linescan performed across the α-Fe2O3 nanorod shows the distinct spatial profiles of Fe and Ni, confirming that Ni species were deposited on the Fe2O3 surface (Fig. S2, ESI†). These results reveal that α-Ni(OH)2 nanoparticles adhere to the surface of α-Fe2O3, forming heterojunctions between the two components.
 | ||
| Fig. 3 (a) HRTEM image of Ni(OH)2/α-Fe2O3 and (b) magnified HRTEM image of the selected frame from image (a). | ||
Fig. 4a shows the linear sweep voltammetric (LSV) curves of the α-Fe2O3 and Ni(OH)2/α-Fe2O3 photoanodes in 1 mol L−1 KOH electrolyte with glucose under chopped light illumination. After loading Ni(OH)2 cocatalyst, the photocurrent for glucose oxidation on the α-Fe2O3 electrode is significantly increased. The Ni(OH)2 film itself shows no photoresponse for glucose oxidation (Fig. S3, ESI†). In 1 mol L−1 KOH electrolyte without glucose, the photocurrent curve of the Ni(OH)2/α-Fe2O3 electrode is different from that of the α-Fe2O3 electrode (Fig. S4, ESI†), which may be because of the oxidation of Ni(OH)2 in the photoelectrochemical process. This suggests that Ni(OH)2 acts as an active site for glucose oxidation.
 | ||
| Fig. 4 (a) LSV curves of Fe2O3 and Ni(OH)2/α-Fe2O3 photoanodes under chopped light illumination; (b) the possible process of glucose oxidation over Ni(OH)2/Fe2O3 photoanode; (c) and (d) LSV curves of α-Fe2O3 photoanodes modified with Fe(OH)3 and Cu(OH)2; reaction condition: 1 mol L−1 KOH aqueous solution with 0.025 mol L−1 glucose; light source: 300 W Xe lamp; scanning rate: 20 mV s−1. | ||
The enhanced photocurrent observed for the Ni(OH)2/α-Fe2O3 sample in Fig. 4b is possibly due to the fact that Ni(OH)2 traps photo-generated holes, forming Ni3+ species, which efficiently catalyzes glucose oxidation while Ni3+ is simultaneously reduced back to Ni2+.29–31 Notably, the cocatalyst Ni(OH)2 can enhance the photo-generated charge separation of α-Fe2O3 and provide catalytic sites for glucose oxidation.
Some metal components (Fe and Cu) with multiple oxidation states were demonstrated to be excellent electrocatalysts for glucose sensors.32,33 Therefore, Fe(OH)3 and Cu(OH)2 were also used to modify the α-Fe2O3 photoanode for glucose oxidation. As shown in Fig. 4c and d, the major phenomenological observation is that the modification of α-Fe2O3 with Fe(OH)3 and Cu(OH)2 can noticeably enhance the photocurrent density. The abovementioned results show that Fe(OH)3 and Cu(OH)2 play a similar role as that of Ni(OH)2. Specifically, they can improve the hole transfer from Fe2O3 to glucose and provide active sites for glucose oxidation.
The α-Fe2O3 based photoanodes with cocatalysts, such as Ni(OH)2, reported in this study can photooxidize various biomass molecules in combination with an O2-reducing cathode. Fig. 5 shows the photocurrent density generated by methanol, glycerol, cellubiose and starch. The photocurrent densities of Ni(OH)2/α-Fe2O3 photoanodes can reach 4.7 and 5.0 mA cm−2 at 0 V vs. SCE fueled by methanol and glycerol, respectively. Whereas cellubiose and starch with relatively high molecular weights give photocurrent densities of approximately 4.0 and 3.5 mA cm−2, respectively, at 0 V vs. SCE with Ni(OH)2/α-Fe2O3 photoanodes. It was noted that the photocurrent density produced by starch was lower than that obtained from cellubiose solution under similar conditions probably because of the slow hydrolysis of starch. The results illustrate that all these biomass materials are promising fuels for PFCs and loading cocatalysts is an efficient method for improving the PFC performance.
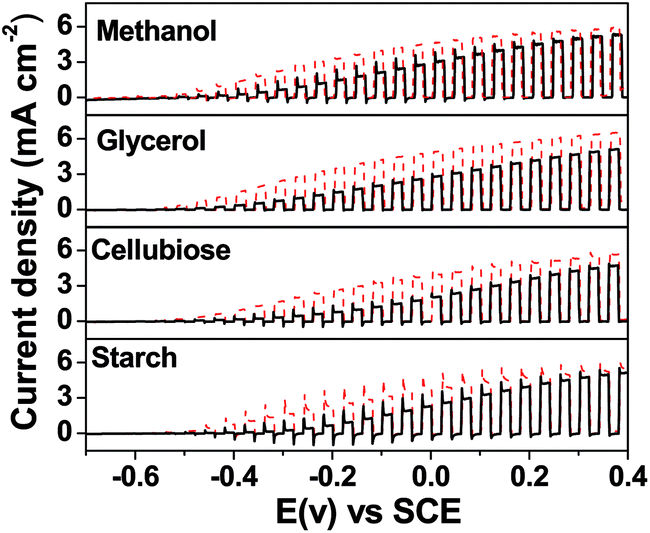 | ||
| Fig. 5 LSV curves of α-Fe2O3 (solid) and Ni(OH)2/α-Fe2O3 (dashed) photoanodes in 1 mol L−1 KOH electrolyte with different biomass-derived compounds under chopped light illumination; methanol (10 vol%); glycerol (0.025 mol L−1); cellubiose (0.025 mol L−1) and starch (1 wt%). Light source: 300 W Xe lamp; scanning rate: 20 mV s−1. | ||
A simple PFC consisting of an α-Fe2O3 (or Ni(OH)2/α-Fe2O3) photoanode and a Pt plate cathode in glucose solution was fabricated. Fig. 6 illustrates the voltage–current density and power density–current density curves, and Table 1 displays the photovoltaic parameters for both the PFCs. The device with the α-Fe2O3 photoanode yields an open-circuit voltage (Voc) of 0.38 V, a short-circuit current (Isc) of 1.17 mA cm−2, and a fill factor (FF) of 0.18, resulting in an overall power conversion efficiency (PCE) of 0.082%. In contrast, the photovoltaic device containing the Ni(OH)2/α-Fe2O3 photoanode presents a Voc of 0.43 V, an Isc of 1.97 mA cm−2 and an FF of 0.21. As a result, the Ni(OH)2/α-Fe2O3 based cell reaches a conversion efficiency of 0.18%, outperforming that of the Fe2O3-based device. As shown in power density–current density curves, the device with the α-Fe2O3 photoanode yields a maximum power density of 0.082 mA cm−2, while the maximum power density (Pmax) of Ni(OH)2/α-Fe2O3 is 0.18 mA cm−2. These results reveal that with an α-Fe2O3 based PFC, biomass can be directly converted into electrical energy and the efficiency can be enhanced by modifying the α-Fe2O3 photoanode with cocatalysts.
 | ||
| Fig. 6 I–V and I–P curves of PFCs for α-Fe2O3 and Ni(OH)2/α-Fe2O3 photoanodes operating with light irradiation in 1 mol L−1 KOH electrolyte containing 0.025 mol L−1 glucose under irradiation with the AM 1.5G simulated solar light (100 mW cm−2). | ||
The stabilities of α-Fe2O3 based photoanodes and the intermediates in the reaction were tested in 1 mol L−1 KOH electrolyte containing glucose. Fig. 7 illustrates the amperometric I–t curves of the α-Fe2O3 and Ni(OH)2/α-Fe2O3 electrodes under continuous illumination at 0 V vs. SCE. The photocurrent of bare α-Fe2O3 after 4 h illumination is 86% of the initial value; however, in the case of the Ni(OH)2/α-Fe2O3 electrode, the photocurrent can be maintained at 96%. Additionally, XPS results (Fig. S5, ESI†) show that the valence state of Ni is maintained during the reaction in 4 h, suggesting that Ni2+ in the Ni(OH)2/α-Fe2O3 electrode can be rapidly regenerated. This suggests that the cocatalyst is rather effective for improving the steady state of the α-Fe2O3 photoanode.
 | ||
| Fig. 7 Amperometric I–t curves of α-Fe2O3 and Ni(OH)2/α-Fe2O3 photoanodes at 0 V vs. SCE in a 1 mol L−1 KOH electrolyte containing 0.025 mol L−1 glucose. Light source: 300 W Xe lamp. The area of α-Fe2O3 film is 1.58 cm2. | ||
The products from glucose with the Ni(OH)2/α-Fe2O3 photoanode were detected by HPLC after 6 h of reaction. These intermediates were identified as arabinose, erythrose, glyceraldehyde, glycolaldehyde, glycolate and formate, and the carbon balance is ca. 71% (Table S1 and Fig. S6, ESI†). The carbon loss of 29% is mainly attributed to the release of CO2, which is produced from the cleavage of the C–C bonds of glucose and its derivatives. This result implies that α-Fe2O3 based photoanodes can efficiently cleave the C–C bond in biomass and oxidize it to CO2 in the PFC at low temperature.
Conclusion
PFCs consisting of an α-Fe2O3 based photoanode and an O2-reducing cathode were fabricated. The sunlight utilization of PFCs was extended to the visible light region. We initially reported that transition metal hydroxides (Ni, Fe and Cu) are excellent cocatalysts of α-Fe2O3 for biomass oxidation. The performance of PFCs can be noticeably enhanced by loading these cocatalysts on an α-Fe2O3 photoanode. Moreover, compared with traditional DAFCs, PFCs can efficiently break the C–C bonds of the biomass, and thus PFCs can be directly powered with natural biomass molecules.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC, grant no. 21090340, 21373209).References
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS.
- J. Potočnik, Science, 2007, 315, 810–811 CrossRef PubMed.
- E. Antolini, ChemSusChem, 2013, 6, 966–973 CrossRef CAS PubMed.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS PubMed.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 97, 1–12 CrossRef CAS PubMed.
- A. Brouzgou, A. Podias and P. Tsiakaras, J. Appl. Electrochem., 2013, 43, 119–136 CrossRef CAS.
- S. D. Minteer, B. Y. Liaw and M. J. Cooney, Curr. Opin. Biotechnol., 2007, 18, 1–7 CrossRef PubMed.
- F. Ahmad, M. N. Atiyeh, B. Pereira and G. N. Stephanopoulos, Biomass Bioenergy, 2013, 56, 179–188 CrossRef CAS PubMed.
- M. Kaneko, J. Nemoto, H. Ueno, N. Gokan, K. Ohnuki, M. Horikawa, R. Saito and T. Shibata, Electrochem. Commun., 2006, 8, 336–340 CrossRef CAS PubMed.
- R. L. Chamousis and F. E. Osterloh, ChemSusChem, 2012, 5, 1–7 CrossRef PubMed.
- P. J. Barczuk, A. Lewera, K. Miecznikowski, P. Kulesza and J. Augustynskiz, Electrochem. Solid-State Lett., 2009, 12, B165–B166 CrossRef CAS PubMed.
- M. Antoniadou, D. Kondarides, D. Labou, S. Neophytides and P. Lianos, Sol. Energy Mater. Sol. Cells, 2010, 94, 592–597 CrossRef CAS PubMed.
- M. Antoniadou and P. Lianos, Catal. Today, 2009, 144, 166–171 CrossRef CAS PubMed.
- Y. Yan, J. Fang, Z. Yang, J. Qiao, Z. Wang, Q. Yu and K. Sun, Chem. Commun., 2013, 49, 8632–8634 RSC.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- D. A. Wheeler, G. Wang, Y. Ling, Y. Li and J. Z. Zhang, Energy Environ. Sci., 2012, 5, 6682–6702 CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048–3055 CrossRef CAS.
- J. Zhu, Z. Yin, D. Yang, T. Sun, H. Yu, H. E. Hoster, H. H. Hng, H. Zhang and Q. Yan, Energy Environ. Sci., 2013, 6, 987–993 CAS.
- S. Saremi-Yarahmadi, K. G. U. Wijayantha, A. A. Tahir and B. Vaidhyanathan, J. Phys. Chem. C, 2009, 113, 4768–4778 CAS.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- Y. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- J. Y. Kim, G. Magesh, D. H. Youn, J. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 Search PubMed.
- H. K. Mulmudi, N. Mathews, X. C. Dou, L. F. Xi, S. S. Pramana, Y. M. Lam and S. G. Mhaisalker, Electrochem. Commun., 2011, 13, 951–954 CrossRef CAS PubMed.
- L. Armelao, R. Bartoncello, L. Crociani, G. Depaoli, G. Granozzi, E. Tondello and M. Benttinelli, J. Mater. Chem., 1995, 5, 79–83 RSC.
- P. R. Martins, M. A. Rocha, L. Angnes, H. E. Toma and K. Araki, Electroanalysis, 2011, 23, 2541–2548 CrossRef CAS.
- J. Nai, S. Wang, Y. Bai and L. Guo, Small, 2013, 9, 3147–3152 CrossRef CAS PubMed.
- S. Xie, T. Zhai, W. Li, M. Yu, C. Liang, J. Gan, X. Lu and Y. Tong, Green Chem., 2013, 15, 2434–2440 RSC.
- X. Cao and N. Wang, Analyst, 2011, 136, 4241–4246 RSC.
- S. Sun, X. Zhang, Y. Sun, S. Yang, X. Song and Z. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 4429–4437 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07498j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |