
Oxygen as single oxidant for two steps: base-free one-pot Pd(ii)-catalyzed alcohol oxidation & arylation to halogen-intact β-aryl α,β-enones†
Abstract
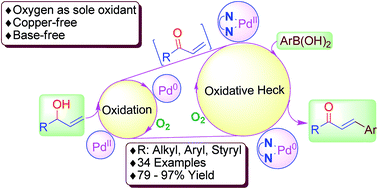
Using oxygen as the sole oxidant for two steps, we developed a new method to synthesize β-aryl α,β-enones by fine-tuning the Pd(II)-catalyzed oxidation of allyl alcohol to subsequent arylation with arylboronic acids, arylboronic ester and aryltrifluoroborate salt. This one-pot green method does not require copper salt, base, and intermediate isolation. Halogen-bearing chalcones, dibenzylideneacetones and arylalkyl enones were synthesized in good yields.
 Please wait while we load your content...
Please wait while we load your content...

