
Diels–Alder reactions of pinacol alkenylboronates: an experimental and theoretical study†
Margarita M. Vallejos,
Nicolás Grimblat and
Silvina C. Pellegrinet*
Instituto de Química Rosario (CONICET), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, Rosario 2000, Argentina. E-mail: pellegrinet@iquir-conicet.gov.ar
First published on 1st August 2014
Abstract
We have studied the Diels–Alder reactions of pinacol alkenylboronates with cyclopentadiene under two different sets of conditions: thermal heating at 170 °C in a pressure tube and with catalytic TFA (5 mol%) at 80 °C. Yields varied significantly from system to system and also for the uncatalyzed and catalyzed methodologies. Moderate to excellent exo-stereoselectivities were obtained in all cases. The theoretical study of the thermal reactions sheds some light on the intriguing substituent effects observed experimentally. A variety of substituted 5-norbornen-2-ols were easily generated by subsequent in situ oxidation of the cycloadducts with alkaline hydrogen peroxide.
Introduction
The Diels–Alder (DA) reactions of boron-activated dienophiles were first described more than five decades ago. In the last few years, a renewed interest in such processes has arisen, both from the experimental and theoretical viewpoints.1–32 We have recently shown that the Diels–Alder reactions of vinylboronates can be easily performed using microwave irradiation giving excellent yields of the cycloadducts. Vinylboronic acid pinacol ester showed good stability towards hydrolysis, operational simplicity and yields of Diels–Alder products. The [4 + 2] cycloadditions of pinacol vinylboronate with a variety of cyclic and acyclic dienes under microwave irradiation generated the boronate cycloadducts in excellent yields in short reaction times (1–6 h) (Scheme 1).32 For example, the reaction with cyclopentadiene was complete in 1 h at 150 °C, affording the products in quantitative yield with a 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :62 endo/exo ratio. Subsequent in situ oxidation of the cycloadducts with alkaline hydrogen peroxide yielded the alcohols efficiently, demonstrating the utility of these intermediates for direct C–O bond-forming reactions.
:62 endo/exo ratio. Subsequent in situ oxidation of the cycloadducts with alkaline hydrogen peroxide yielded the alcohols efficiently, demonstrating the utility of these intermediates for direct C–O bond-forming reactions.
 | ||
| Scheme 1 | ||
As part of our continuing work in the field, we have now studied the Diels–Alder reactions of cyclopentadiene with pinacol alkenylboronates with different substitution patterns under different reaction conditions with the aims of developing new methodologies, gaining additional knowledge about the reactivity of boron-substituted dienophiles and analyzing their possible use as synthetic equivalents of substituted enols.
Results and discussion
To carry out this study we have used cyclopentadiene, which was chosen for being a reactive cyclic 1,3-diene and also for the interesting structural and synthetic properties of the bicyclo[2.2.1]heptane products.33 To investigate the substituent effect on the outcome of the thermal Diels–Alder reaction, we tested a range of commercially available alkenylboronates with alkyl or aryl groups with different substitution patterns in the 1- and 2-positions of the carbon–carbon double bond. Initial screening reactions under microwave heating with the pinacol esters of trans-1-penten-1-ylboronic acid and trans-2-phenylvinylboronic acid suggested that the presence of substituents in the double bond of the substrates retarded the cycloaddition process considerably. Therefore, the use of microwave irradiation proved impractical. We then re-investigated the Diels–Alder reaction of pinacol vinylboronate with cyclopentadiene under a large number of thermal conditions using conventional heating. Table 1 summarizes the outcome of some descriptive experiments. Entry 1 shows the result previously obtained in our laboratories at 150 °C under microwave irradiation for 1 h.32 When the reaction was performed in refluxing xylenes under conventional heating, a 54% yield was obtained (entry 2). We managed to get a 96% yield in refluxing toluene with a longer reaction time (entry 3). As an alternative, use of a pressure tube at 150 °C in 1 h gave the cycloadduct in 79% yield (entry 4), while increasing the time to 2 h raised the yield to 86% (entry 5). If the bath temperature was set to 170 °C, a 92% yield was generated in 5 h, and a nearly quantitative yield was obtained in 1 h (entries 6 and 7). BHT (5 mol%) was added to prevent undesired radical side reactions. The endo/exo ratios varied slightly around 38:62 for all reactions.
|
||
|---|---|---|
| Entry | Conditions | Yield (%) endo/exoa |
| a Determined by 1H NMR. | ||
| 1 | Toluene, 150 °C, MW, 1 h32 | 100 |
| 38:62 |
||
| 2 | Xylenes, reflux, 1 h | 54 |
| 35:65 |
||
| 3 | Toluene, reflux, 5 h | 96 |
| 35:65 |
||
| 4 | Toluene, 150 °C, 1 h, pressure tube | 79 |
| 32:68 |
||
| 5 | Toluene, 150 °C, 2 h, pressure tube | 86 |
| 37:63 |
||
| 6 | Toluene, 170 °C, 5 h, pressure tube, BHT (5 mol%) | 92 |
| 40:60 |
||
| 7 | Toluene, 170 °C, 1 h, pressure tube, BHT (5 mol%) | 96 |
| 35:65 |
||
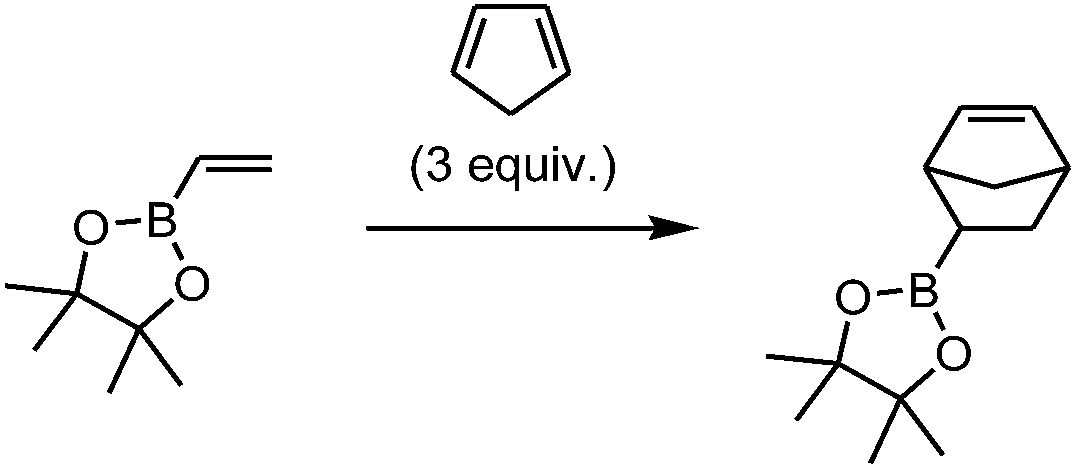
Having optimized the conditions for the thermal reaction of pinacol vinylboronate under conventional heating, we next turned our attention to the [4 + 2] cycloadditions of the substituted substrates (Table 2). All the reactions were optimized to yield the greatest amount of products. Lower temperatures or shorter reaction times afforded poorer yields while higher temperatures or longer reaction times either did not increase the yield or led to some decomposition.
|
||||
|---|---|---|---|---|
| Entry | Dienophile | Time (h) | Yielda (%) endo/exob | Products |
| a Yields based on recovered starting materials (BRSM) in parenthesis.b Relative to the pinacol boronate moiety, determined by 1H NMR.c The starting material was recovered. | ||||
| 1 |  |
1 | 96 | 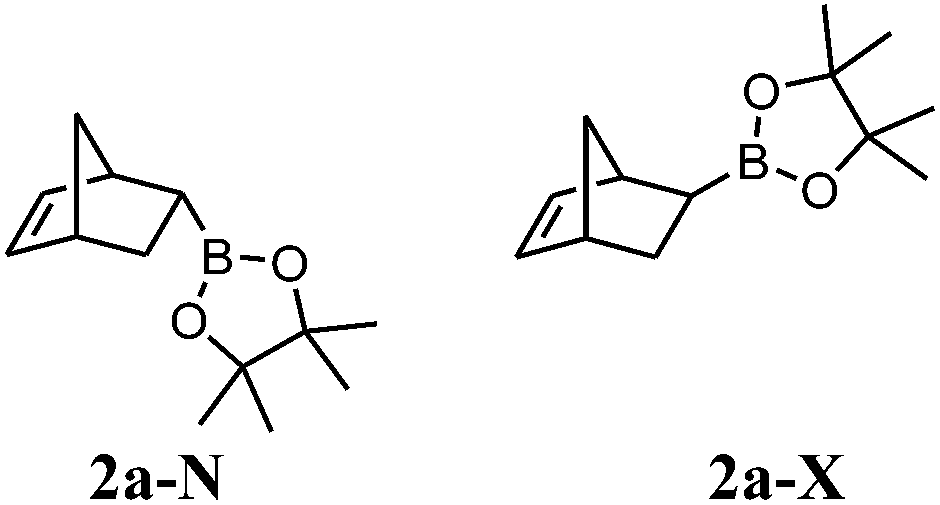 |
| 35:65 |
||||
| 2 |  |
24 | 78 (100) |  |
| 17:83 |
||||
| 3 | 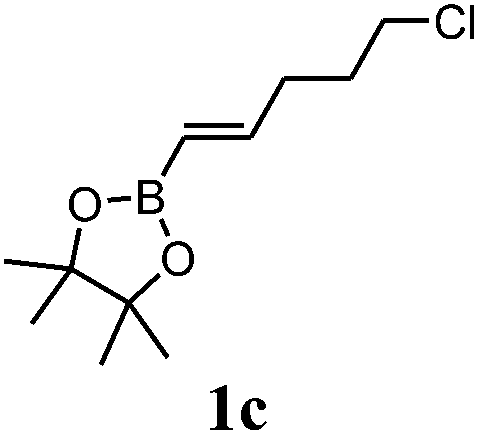 |
24 | 89 |  |
| 20:80 |
||||
| 4 | 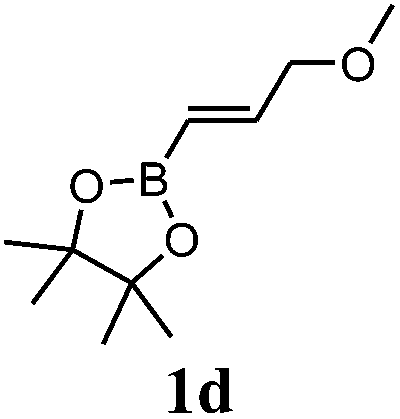 |
24 | 88 (100) |  |
| 15:85 |
||||
| 5 |  |
24 | 29 (90) |  |
| 35:65 |
||||
| 6 |  |
24 | 20 (70) |  |
| 40:60 |
||||
| 7 |  |
24 | 19 (48) |  |
| 37:63 |
||||
| 8 |  |
1 | 25 (91) | 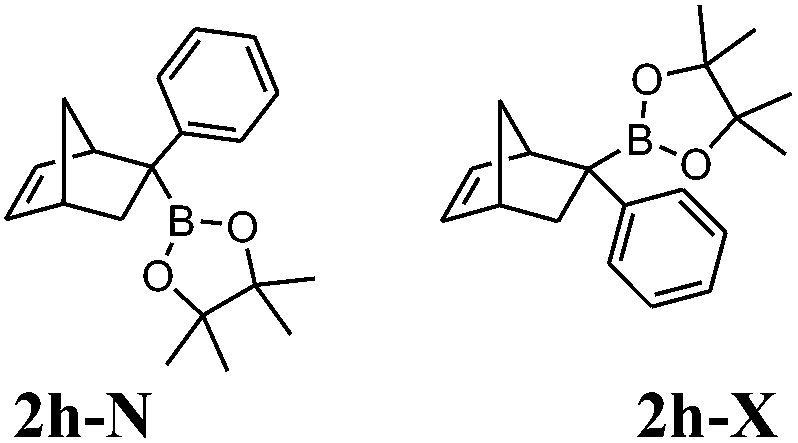 |
| 10:90 |
||||
| 9 |  |
24 | 72 (80) |  |
| 9:91 |
||||
| 10 |  |
12 | Tracesc |  |
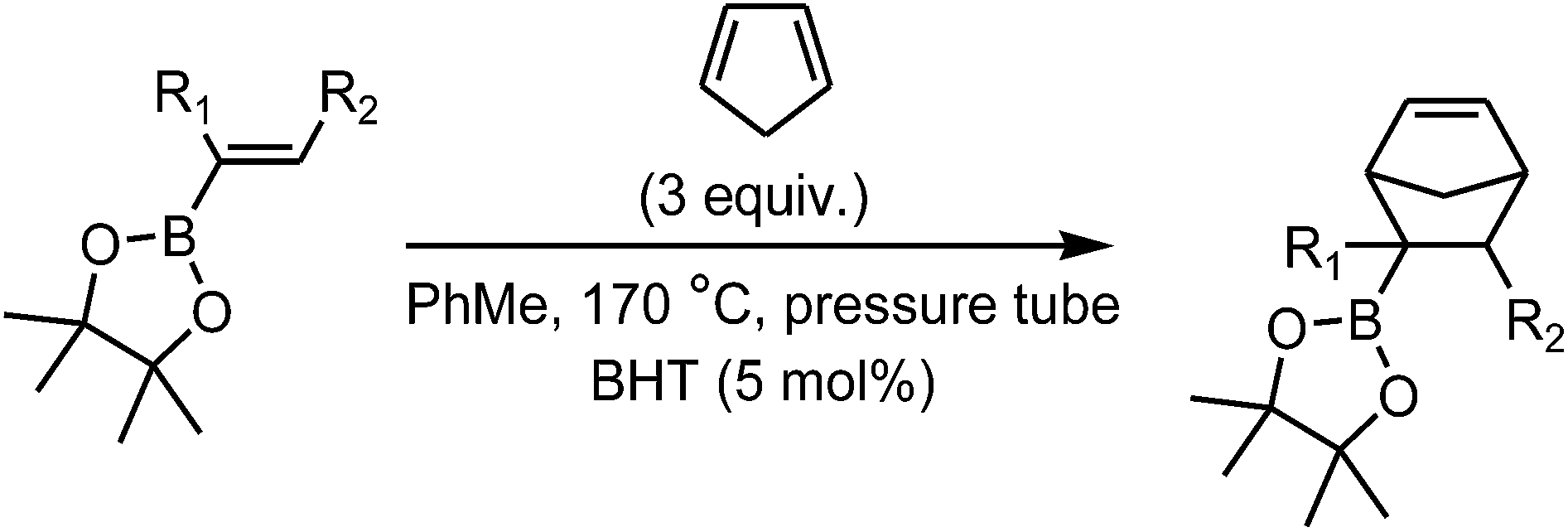
As found in our initial experiments with microwave heating, longer reactions times than for the parent dienophile (1a) were needed in all cases, excluding 1h (entry 8). However, for the latter the yield was very low and did not increase by extending the reaction time (25% in 1 h endo/exo 10:90, 21% in 12 h endo/exo 41:59). Alkyl-substituted substrates performed much better than the aromatic analogues, giving yields in the range 72–89% (entries 2–4 and 9). We reasoned that the conjugated aromatic ring donated electron density to the carbon–carbon double bond. However, the introduction of electron-withdrawing substituents on the phenyl ring did not improve the reactivity of such systems (entries 5–8 and 10). Regarding the stereoselectivities, the exo cycloadduct predominated in all reactions. The highest exo-stereoselectivity was observed for isopropenylboronic acid pinacol ester (1i) (endo/exo 9:91, entry 9), while alkenylboronates with alkyl groups in the 2-position exhibited endo/exo ratios higher than 20:80 (entries 2–4). The aromatic compounds showed moderate exo-selectivities, similar to the one obtained with the unsubstituted system (endo/exo ∼ 40:60).
In the next stage, we aimed to determine whether milder conditions could be used so we embarked in the development of the acid-catalyzed version of the reaction under study (Table 3).34–37 Many experiments were run for the Diels–Alder reactions of pinacol esters of vinylboronic acid (1a), trans-1-penten-1-ylboronic acid (1b) and trans-2-phenylvinylboronic acid (1e) to determine the optimal conditions. Brønsted acids gave better results than Lewis acids, due to the greater polymerization of the diene in the presence of the latter. Among the Brønsted acids, we tried acetic acid, trifluoroacetic acid (TFA) and triflic acid. We tested up to 2 equivalents of Brønsted acids and 10 equivalents of cyclopentadiene, solvents like toluene, dichloromethane and water, and temperatures ranging from room temperature to 150 °C. For the reaction of 1a, we determined that best yields of the cycloadducts were obtained in 5 h at 80 °C with 5 mol % of TFA (entry 1, 88%, endo/exo 36:64). When we run the reaction in the absence of TFA, a 20% yield was generated, with the same endo/exo ratio. Use of a pressure tube, though not necessary in toluene at 80 °C, was preferred to avoid evaporation of the small loading of the catalyst (Bp 72.4 °C). Quite unexpectedly, under catalyzed conditions only 1-phenylvinylboronic acid pinacol ester (1h) performed well (entry 8, 83%, endo/exo 6:94). The other dienophiles gave yields below 45%. However, it is interesting to note that in this case aromatic alkenylboronates afforded better yields than the aliphatic compounds. Also, considerably higher exo-selectivities than for the uncatalyzed reactions were observed. Possibly, the acid catalyst interacts with the π electrons of the aromatic ring and therefore withdraws electron density from the conjugated unsaturated system leading to the activation of the double bond. Within the aliphatic alkenylboronates, 1d, having a possible site for protonation (oxygen atom) gave better results than 1b and 1c.
|
||||
|---|---|---|---|---|
| Entry | Dienophile | Time (h) | Yielda (%) endo/exob | Products |
| a Yields based on recovered starting materials (BRSM) in parenthesis.b Relative to the pinacol boronate moiety, determined by 1H NMR.c The starting material was recovered. | ||||
| 1 |  |
5 | 88 |  |
| 36:64 |
||||
| 2 |  |
24 | Tracesc |  |
| 3 |  |
24 | Tracesc |  |
| 4 |  |
24 | 17 (75) |  |
| 14:86 |
||||
| 72 | 26 (68) | |||
| 10:90 |
||||
| 5 |  |
24 | 32 (92) |  |
| 17:83 |
||||
| 72 | 38 (98) | |||
| 17:83 |
||||
| 6 |  |
24 | 20 (70) |  |
| 36:64 |
||||
| 72 | 31 (70) | |||
| 29:71 |
||||
| 7 |  |
24 | 24 (78) |  |
| 10:90 |
||||
| 72 | 45 (73) | |||
| 10:90 |
||||
| 8 |  |
12 | 83 |  |
| 6:94 |
||||
| 9 |  |
24 | 15 (24) |  |
| 9:91 |
||||
| 10 |  |
12 | Tracesc |  |

We were surprised to note that the background reaction of dienophile 1h afforded a very high yield of the corresponding boronate cycloadduct (91%) with excellent exo-selectivity (endo/exo 5:95) (Scheme 2). Under the same conditions, vinylboronic acid pinacol ester (1a) gave a lower yield (45%), which was a bit unexpected since previous experiments at higher temperatures suggested that the parent compound was more reactive than 1h (Table 2).
 | ||
| Scheme 2 | ||
We tested whether we could perform the catalyzed reaction of alkenylboronate 1h at room temperature using the same amount of diene, catalyst, and BHT, but we only obtained a 17% yield with a 3:97 endo/exo ratio (96% BRSM) after 12 h.
Since, as commented above, prolonged exposure to the reaction conditions did not increase the yields of the products we figured that thermodynamic equilibria had been reached. Also, dienophile 1h gave a 25% (91% BRSM) in 1 h at 170 °C (entry 8, Table 2), while the yield was much better after 12 h at 80 °C (91%, Scheme 2), so we suspected that under the initial thermal conditions the energy barrier of the Diels–Alder reaction has been surpassed and that some retro Diels–Alder might have taken place. For that reason, we submitted cycloadduct 2g (endo/exo 10:90) to the conditions of the uncatalyzed thermal reaction (Scheme 3). Indeed, the retro Diels–Alder reaction occurred, giving 76% of alkenylboronate 1g and 15% of recovered cycloadduct 2g (a mixture with a very similar composition to the one obtained when submitting the direct reaction).
 | ||
| Scheme 3 | ||
Finally, we studied the tandem Diels–Alder reaction of alkenylboronates with cyclopentadiene-oxidation (Table 4).
|
|||
|---|---|---|---|
| Entry | Dienophile | Overall yield (%) endo/exoa | Products DA + [O] |
| a Determined by NMR integration.b Non TFA-catalyzed thermal conditions were used.c TFA-catalyzed conditions were used.d Conditions shown in Scheme 2 were used. | |||
| 1 |  |
93b |  |
| 39:61 |
|||
| 2 |  |
79b |  |
| 15:85 |
|||
| 3 |  |
82b |  |
| 14:86 |
|||
| 4 |  |
82b |  |
| 12:88 |
|||
| 5 |  |
37c |  |
| 13:87 |
|||
| 6 |  |
28c |  |
| 13:87 |
|||
| 7 |  |
44c |  |
| 9:91 |
|||
| 8 |  |
88d |  |
| 3:97 |
|||
| 9 |  |
66b |  |
| 9:91 |
|||

Except for the compounds with aryl groups at the 2-position (entries 5–7), we coupled the non-catalyzed thermal conditions shown in Table 2 for the cycloaddition step with the final oxidation with alkaline hydrogen peroxide in one-pot. Overall yields for the two-step sequence were very similar to the ones obtained in the Diels–Alder reactions, which suggests that in situ transformation of the boronate cycloadducts to the corresponding alcohols occurs very efficiently. In general, the substituted 5-norbornen-2-ols were obtained with acceptable to very good yields, which demonstrated that alkenylboronic esters can be used as synthetic equivalents of substituted enols. Due to their high functionalization, the alcohol products can be foreseen as valuable synthetic intermediates towards a variety of chemical structures. We anticipate that other transformations of the cycloadduct intermediates could be developed for further elaboration of C–C, C–O and C–N bonds.
Computational study
To gain a deeper insight into the mechanism of the Diels–Alder reactions of the dienophiles under study we performed a theoretical study. In particular, we intended to examine the reversibility of such processes and whether the starting material/products distribution was determined by thermodynamic or kinetic control. Therefore, we optimized the geometries in toluene of the reactants, the transition structures and the products to compute the activation and reaction energies at 170 °C. In addition, we analyzed the geometries and the properties of the dienophiles and the transition structures with the aim of rationalizing the reactivity and selectivity trends.Computational methods
All calculations were performed with the Gaussian 09 package.38 We carried out thorough conformational analyses to locate the lowest energy geometry for all the structures under study. Final geometry optimizations were carried out using MPWB1K global-hybrid meta-GGA functional39 together with 6-311G* basis. Solvent effects of toluene were taken into account through full optimizations using the polarizable continuum model (PCM) as developed by Tomasi's group40 in the framework of self-consistent reaction field (SCRF).41–43 The vibrational frequencies were calculated to determine the nature of the stationary points and to evaluate zero-point vibrational energy (ZPVE) and thermal corrections at 443 K (170 °C). The frontier molecular orbitals (FMOs) were computed with the same method. Intrinsic Reaction Coordinate (IRC) calculations were run to verify if the transition structures were directly connected to the reactants and the products.Fig. 1 shows the free energy profiles for the Diels–Alder reactions of selected dienophiles with cyclopentadiene as a means to compare the reaction channels (for all the energy profiles see the ESI†). Also, the optimized geometries for the corresponding transition structures with selected distances and Wiberg bond indexes are shown. Table 5 gathers the computed free energies of activation, reaction free energies and endo/exo selectivities at 170 °C in toluene for all the Diels–Alder reactions under study. All transition structures exhibit classical [4 + 2] geometries and are asynchronic. However, the ones corresponding to the dienophiles with aromatic substituents in C-2 (1e–1g) are less asynchronic and the asynchronicity is reversed, i.e. the carbon atom directly attached to boron (C-1) is closer to the diene carbon than C-2. Carbon–carbon distances for the other systems are in line with previous results: the presence of the boron atom makes C-2 more electron deficient, so it becomes closer to the corresponding carbon atom in the diene than C-1.44 Analysis of FMOs indicates that the reactions under study are normal electron-demand Diels–Alder reactions. From the atomic coefficients for the LUMOs of the dienophiles, it appears that the computed reversal of asynchonicity is caused by electronic effects since compounds 1e–1g have larger coefficients at C-1 than at C-2, in contrast to the rest of the dienophiles. However, we do not discard the contribution of steric effects. In addition, the transition structures of alkenylboronates with aromatic susbtituents in C-1 (1h and 1j) are extremely asynchronic, with asynchronicities as high as 0.66 Å. Nonetheless, IRC calculations connected the transition structures with the reactants and the products, therefore all reactions were computed to be concerted. In this case, comparison of the atomic coefficients corresponding to the LUMOs of the dienophiles suggests that the higher asynchronicity is determined by steric effects rather than electronic effects. The non-classical [4 + 3] carbon–boron interactions are weak (C–B distances 2.70–3.15 Å, WBI 0.04-002) and very similar for the endo and exo approaches. Consequently, the observed moderate to high exo-selectivities seems to be a consequence of unfavorable van der Waals interactions in the endo transition structures.
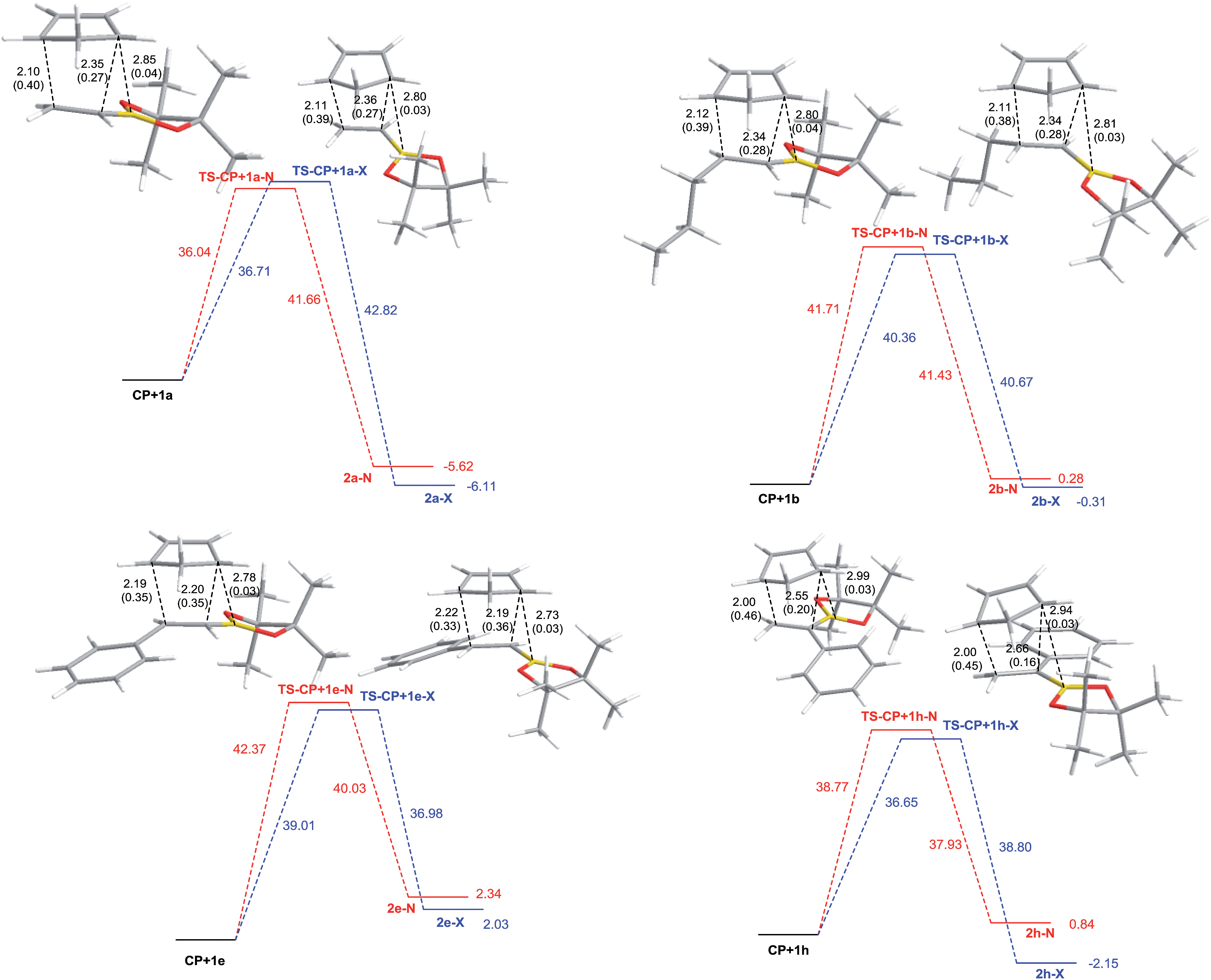 | ||
| Fig. 1 MPWB1K/6-311G* free energy profiles for the Diels–Alder reactions of pinacol alkenylboronates 1a (top left), 1b (top right), 1e (bottom left) and 1h (bottom right) with cyclopentadiene (free activation energies in toluene at 170 °C for the direct and reverse reaction, in kcal mol−1). The optimized geometries in toluene for the transition structures with selected distances in Å and Wiberg bond indexes in parentheses are also shown. | ||
| Dienophile | TS | ΔG#Tol | endo/exo | ΔGTol | endo/exo |
|---|---|---|---|---|---|
| a Energies in kcal mol−1. | |||||
| 1a | endo | 36.04 | 68:32 |
−5.62 | 37:63 |
| exo | 36.71 | −6.11 | |||
| 1b | endo | 41.71 | 18:82 |
0.28 | 34:66 |
| exo | 40.36 | −0.31 | |||
| 1c | endo | 41.25 | 17:83 |
−1.09 | 61:39 |
| exo | 39.81 | −0.70 | |||
| 1d | endo | 39.44 | 10:90 |
−3.99 | 46:54 |
| exo | 37.53 | −4.15 | |||
| 1e | endo | 42.37 | 2:98 |
2.34 | 41:59 |
| exo | 39.01 | 2.03 | |||
| 1f | endo | 40.75 | 29:71 |
1.36 | 79:21 |
| exo | 39.97 | 2.53 | |||
| 1g | endo | 40.72 | 22:78 |
2.16 | 51:49 |
| exo | 39.59 | 2.19 | |||
| 1h | endo | 38.77 | 8:92 |
0.84 | 3:97 |
| exo | 36.65 | −2.15 | |||
| 1i | endo | 40.25 | 8:92 |
−1.30 | 51:49 |
| exo | 38.13 | −1.24 | |||
| 1j | endo | 41.92 | 24:76 |
1.47 | 79:21 |
| exo | 40.91 | 2.65 | |||
Also, the short distance (ca. 2.4 Å) between one of the methylene hydrogens of the cyclopentadiene moiety and one of the oxygens of the pinacol boronate in the exo transition structures suggests the possibility that hydrogen bond interactions contribute to determine the diastereoselectivity. NBO calculations indicate that this accounts for a stabilization of 0.30–0.75 kcal mol−1 of the exo transition structures relative to their endo counterparts.
The lowest energy barriers correspond to the reactions of substrates 1a and 1h, while the one for analogue 1j is the highest one, in accordance with the experimental reactivities. However, the free energies of activation for the other reactions do not match the reactivity trend accurately. For that reason, we optimized the geometries of the products and computed the reaction energies. By analyzing the barriers for the direct reactions (Diels–Alder reaction) and the reverse reaction (retro Diels–Alder reaction) we propose that the low product yields for the reactions of dienophiles 1e–1g at 170 °C, might be related to the higher reversibility of the reactions as a result of the higher energies of the products and the resulting lower energy barriers for the retro Diels–Alder reactions. The higher energies of the products corresponding to the reactions of aromatic alkenylboronates 1e–1g appear to be originated from steric clashes between the aromatic ring and the [2.2.1] backbone.
For 1b–1d and 1i, the endo/exo selectivities calculated from activation free energies are in agreement with the experimental values. For the more reactive dienophiles 1a and 1h the calculated endo/exo selectivities from the reaction energies are in excellent accordance with the experimental outcome, indicating the dominance of the thermodynamical control in these reactions. The endo/exo ratios for 1e–1g are closer to the figures obtained from reaction energies, which supports that in these cases the starting material/products distribution is a consequence of thermodynamic equilibration. On the other hand, free reaction energies of the products predict that the reactions with the 1a–1d and 1h–1i should be exergonic and therefore, the boronate cycloadducts should predominate while that the reactions with 1e–1g and 1j should be endergonic and the starting alkenylboronates should be the major components of the reaction mixtures. Therefore, free energy trends, gave us a hint to better understand the reaction mechanism.
Another point that deserves to be remarked is the high reactivity of substrate 1h. Inspection of the geometry of the corresponding transition structures reveals that a non-classical hydrogen bond (NCHB) between an aromatic proton at the ortho position and one of the oxygens of the pinacol boronate might be responsible for the peculiar reactivity. Such interaction is much stronger in the exo transition structure than in its endo counterpart (exo: 2.17 Å, 1.25 kcal mol−1, endo: 2.38 Å, 0.28 kcal mol−1), and also than in the starting dienophile (2.55 Å, 0.15 kcal mol−1). The unexpected lack of reactivity of structurally related analogue 1j is reflected in a higher free energy barrier obtained from the calculations, which might result from geometric constraints imposed by the bulky chlorine atom. The dihedral angle between the aromatic ring and the double bond in the optimized geometry of the reactant is 54 degrees, making the approach of the diene more difficult. The aforementioned dihedral angle is reduced to roughly 26 degrees in the transition structures, in contrast to the planar geometries corresponding to 1-phenylvinylboronic acid pinacol ester (1h).
Conclusions
We have investigated the Diels–Alder reactions of pinacol alkenylboronates with cyclopentadiene. The outcome of the studied transformation was shown to be very sensitive to the substitution of the dienophile both under thermal and TFA-catalyzed conditions. Theoretical calculations disclosed some interesting substituent effects for these [4 + 2] cycloadditions. We have found that the thermal Diels–Alder reactions of alkenylboronates with aryl groups in the 2-position give low yields because they are highly reversible. The high reactivity of 1-phenylvinylboronic acid pinacol ester (1h) was explained in terms of a stabilizing non-classical hydrogen bond between an aromatic proton and the boronate moiety. We have also synthesized a range of substituted 5-norbornen-2-ols in one-pot by performing the tandem Diels–Alder reactions – alkaline hydrogen peroxide oxidation, demonstrating the versatility of alkenylboronic esters as synthetic equivalents of substituted enols.Experimental section
General experimental procedures
All reagents and solvents were used directly as purchased or purified according to standard procedures. Analytical thin layer chromatography was carried out using commercial silica gel plates (Merck, Silica Gel 60 F254) and visualization was effected with short wavelength UV light (254 nm) and a p-anisaldehyde solution (2.5 mL of p-anisaldehyde + 2.5 mL of H2SO4 + 0.25 mL of AcOH + 95 mL of EtOH). Column chromatography was performed with silica gel 60 H (Merck), slurry packed, run under low pressure of nitrogen. The Diels–Alder reactions were monitored using TLC and 11B NMR analysis in CDCl3. NMR spectra were recorded at 300 MHz for 1H, 75 MHz for 13C, 96 MHz for 11B and 282 MHz for 19F NMR on a Bruker Avance-300 DPX spectrometer with CDCl3 as solvent and (CH3)4Si (1H) and CDCl3 (13C, 76.9 ppm) as internal standards. 11B and 19F NMR spectra were externally referenced to BF3–Et2O and CFCl3, respectively. Chemical shifts are reported in delta (δ) units in parts per million (ppm) and splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad. Coupling constants are recorded in Hertz (Hz). Isomeric ratios were determined by 1H NMR integration. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrometer using sodium chloride plates or potassium bromide pellets. Absorbance frequencies are recorded in reciprocal centimeters (cm−1). The high resolution mass spectra (HRMS) were obtained with a Bruker MicroTOF-Q II instrument (Bruker Daltonics, Billerica, MA). Detection of the ions was performed with electrospray ionization (ESI), positive ion mode and Atmospheric Pressure Chemical Ionization (APCI). The structure of the products were determined by a combination of spectroscopic methods such as IR, 1D and 2D NMR (including NOE, DEPT, COSY, HSQC and HMBC experiments) and HRMS. In some cases, NMR calculations were also performed to corroborate the stereochemistry and the assignment. In addition, we confirmed the structure of the Diels–Alder products by oxidation of the boronates to the alcohols, some of which were described in the literature.Diels–Alder reactions of alkenylboronates: synthesis of boronates 2a–2i
(a) Procedure A: reaction time: 1 h. Yield: 96% (59.1 mg), endo/exo 36:65.
(b) Procedure B: reaction time: 5 h. Yield: 88% (54.2 mg), endo/exo 36:64.
:83.
Boronate 2b-X (major compound, yellowish oil). IR (film) νmax 2956, 2926, 2870, 2359, 2344, 1371, 1312, 1146, 978, 853, 698 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.17 (dd, J5,6 = 5.6, J1,6 = 2.9 Hz, 1H, H-6), 5.87 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 1H, H-5), 2.77 (br s, 1H, H-4), 2.74 (br s, 1H, H-1), 2.15–2.04 (m, 1H, H-3), 1.36–1.26 (m, 4H, H-7 and H-11), 1.24 (br s, 12H, H-9), 1.10–0.92 (m, 2H, H-10), 0.85 (t, J11,12 = 7.3 Hz, 3H, H-12), 0.16 (dd, J2,3 = 5.3, J1,2 = 1.6 Hz, 1H, H-2). 13C NMR (75 MHz, CDCl3) δ 138.3 (CH, C-6), 131.5 (CH, C-5), 82.8 (2C, C-8), 48.5 (CH2, C-7), 46.1 (CH, C-4), 45.0 (CH, C-1), 42.2 (CH, C-3), 37.6 (CH2, C-10), 24.7 (2CH3, C-9), 24.6 (2CH3, C-9), 21.8 (CH2, C-11), 14.4 (CH3, C-12), C-2 signal missing. 11B NMR (96 MHz, CDCl3) δ 34.2.
Boronates 2b-X and 2b-N (yellowish oil). IR (film) νmax 2957, 2926, 2870, 2359, 2342, 1371, 1312, 1244, 1146, 968, 853, 692 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.17 (dd, J5,6 = 5.6, J1,6 = 2.9 Hz, 1H, H-6X), 6.10 (dd, J5,6 = 5.5, J4,5 = 3.1 Hz, 1H, H-5N), 5.98 (dd, J5,6 = 5.5, J4,5 = 2.8 Hz, 1H, H-6N), 5.87 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 1H, H-5X), 2.92 (br s, 1H, H-1N), 2.77 (br s, 1H, H-4X), 2.74 (br s, 1H, H-1X), 2.50 (br s, 1H, H-4N), 2.15–2.04 (m, 1H, H-3X), 1.40–1.26 (m, 11H, H-7X, H-11X, H-3N, H-7N, H-10N and H-11N), 1.24 (s, 12H, H-9X), 1.18 (s, 12H, H-9N), 1.10–0.92 (m, 2H, H-10X), 0.92–0.78 (m, 4H, H-2N and H-12N), 0.85 (t, J11,12 = 7.3 Hz, 3H, H-12X), 0.16 (dd, J2,3 = 5.3, J1,2 = 1.6 Hz, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 138.3 (CH, C-6X), 137.2 (CH, C-5N), 135.1 (CH, C-6N), 131.5 (CH, C-5X), 82.7 (2C, C-8N), 82.2 (2C, C-8X), 48.5 (CH2, C-7X), 47.2 (CH2, C-7N), 47.1 (CH, C-4N), 46.1 (CH, C-4X), 45.0 (CH, C-1X), 44.7 (CH, C-1N), 42.2 (CH, C-3X), 42.0 (CH, C-3N), 39.6 (CH2, C-10N), 37.6 (CH2, C-10X), 24.8 (2CH3, C-9N), 24.7 (2CH3, C-9X), 24.6 (2CH3, C-9X), 24.5 (2CH3, C-9N), 21.9 (CH2, C-11N), 21.8 (CH2, C-11X), 14.4 (2CH3, C-12X and C-12N), C-2 signals missing. 11B NMR (96 MHz, CDCl3) δ 34.1. HRMS (APCI) calcd for C16H28BO2 (M + H)+ 263.2177, found 263.2178.
:80.
Boronate 2c-X (major compound, yellowish oil). IR (film) νmax 2965, 2926, 2358, 2341, 1373, 1314, 1144, 852, 669, 430, 411 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.20 (dd, J5,6 = 5.3, J1,6 = 3.2 Hz, 1H, H-6), 5.88 (dd, J5,6 = 5.3, J4,5 = 2.9 Hz, 1H, H-5), 3.51 (t, J11,12 = 6.9 Hz, 2H, H-12), 2.78 (br s, 2H, H-1 and H-4), 2.14–2.04 (m, 1H, H-3), 1.75 (quintet, J10,11 = J11,12 = 7.2 Hz, 2H, H-11), 1.34–1.27 (m, 2H, H-7), 1.24 (s, 12H, H-9), 1.16–1.02 (m, 2H, H-10), 0.18 (br d, J2,3 = 5.2 Hz, 1H, H-2). 13C NMR (75 MHz, CDCl3) δ 138.6 (CH, C-6), 131.2 (CH, C-5), 83.0 (2C, C-8), 48.6 (CH2, C-7), 46.2 (CH, C-1), 45.3 (CH2, C-12), 45.0 (CH, C-4), 41.6 (CH, C-3), 32.5 (CH2, C-10), 31.8 (CH, C-11), 24.7 (4CH3, C-9), C-2 signal missing. 11B NMR (96 MHz, CDCl3) δ 33.8.
Boronates 2c-X and 2c-N (yellowish oil). IR (film) νmax 2965, 2930, 2358, 2342, 1373, 1144, 852, 717, 546, 411, 401 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.20 (dd, J5,6 = 5.3, J1,6 = 3.2 Hz, 1H, H-6X), 6.17 (dd, J5,6 = 5.6, J4,5 = 3.2 Hz, 1H, H-5N), 6.00 (dd, J5,6 = 5.6, J1,6 = 2.8 Hz, 1H, H-6N), 5.88 (dd, J5,6 = 5.3, J4,5 = 2.9 Hz, 1H, H-5X), 3.58 (t, J11,12 = 6.8, 1H, H-12N), 3.57 (t, J11,12 = 6.9, 1H, H-12N), 3.51 (t, J11,12 = 6.9 Hz, 2H, H-12X), 2.95 (br s, 1H, H-1N), 2.78 (br s, 2H, H-1X and H-4X), 2.51 (br s, 1H, H-4N), 2.14–2.04 (m, 1H, H-3X), 1.84 (quintet, J10,11 = J11,12 = 7.0 Hz, 2H, H-11N), 1.75 (quintet, J10,11 = J11,12 = 7.2 Hz, 2H, H-11X), 1.44–1.41 (m, 1H, H-3N), 1.38–1.32 (m, 2H, H-7N), 1.34–1.27 (m, 2H, H-7X), 1.24 (s, 12H, H-9X), 1.18 (br s, 12H, H-9N), 1.16–1.02 (m, 4H, H-10X and H-10N), 0.84–0.79 (m, 1H, H-2N), 0.18 (br d, J2,3 = 5.2 Hz, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 138.7 (CH, C-6X), 137.1 (CH, C-5N), 135.4 (CH, C-6N), 131.2 (CH, C-5X), 83.0 (2C, C-8X), 82.8 (2C, C-8N), 48.6 (CH2, C-7X), 47.2 (CH2, C-7N and CH, C-4N), 46.2 (CH, C-4X), 45.3 (2CH2, C-12X and C-12N), 45.0 (CH, C-1X), 44.6 (CH, C-1N) 41.6 (CH, C-3X), 41.0 (CH, C-3N), 32.5 (2CH2, C-10X and C-10N), 31.8 (2CH2, C-11X and C-11N), 24.8 (2CH3, C-9N), 24.7 (4CH3, C-9X), 24.5 (2CH3, C-9N), C-2 signals missing. 11B NMR (96 MHz, CDCl3) δ 34.0. HRMS (APCI) calcd for C16H27BClO2 (M + H)+ 297.1787, found 297.1822.
(a) Procedure A: reaction time: 24 h. Yield: 88% (51.1 mg), endo/exo 15:85.
(b) Procedure B: reaction time: 72 h. Yield: 26% (15.1 mg), endo/exo 10:90.
Boronate 2d-X (major compound, yellowish oil). IR (film) νmax 3055, 2976, 2926, 2868, 1406, 1369, 1313, 1145, 1109, 852, 723 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.18 (dd, J5,6 = 5.6, J1,6 = 3.0 Hz, 1H, H-6), 5.90 (dd, J5,6 = 5.6, J4,5 = 3.0 Hz, 1H, H-5), 3.29 (s, 3H, H-11), 3.14 (dd, J10a,10b = 9.5, J3,10a = 5.7 Hz, 1H, H-10a), 2.93 (br s, 1H, H-4), 2.84 (t, J10a,10b = J3,10b = 9.5 Hz, 1H, H-10b), 2.79 (br s, 1H, H-1), 2.42 (m, 1H, H-3), 1.33 (br s, 2H, H-7), 1.23 (s, 12H, H-9), 0.08 (br d, J2,3 = 5.9 Hz, 1H, H-2). 13C NMR (75 MHz, CDCl3) δ 138.4 (CH, C-6), 131.5 (CH, C-5), 83.0 (2C, C-8), 76.5 (CH2, C-10), 58.6 (CH3, C-11), 48.4 (CH2, C-7), 44.4 (CH, C-1), 44.2 (CH, C-4), 41.9 (CH, C-3), 24.7 (4CH3, C-9), C-2 signal missing. 11B NMR (96 MHz, CDCl3) δ 34.0. HRMS (APCI) calcd for C15H26BO3 (M + H)+ 265.1970, found 265.1967.
Boronates 2d-X and 2d-N (yellowish oil). IR (film) νmax 3055, 2976, 2927, 2889, 2868, 1371, 1313, 1145, 1107, 974, 852, 723 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.18 (dd, J5,6 = 5.6, J1,6 = 3.0 Hz, 1H, H-6X), 6.10 (dd, J5,6 = 5.6, J4,5 = 3.1 Hz, 1H, H-5N), 6.04 (dd, J5,6 = 5.5, J1,6 = 2.8 Hz, 1H, H-6N), 5.90 (dd, J5,6 = 5.6, J4,5 = 3.0 Hz, 1H, H-5X), 3.49 (dd, J10a,10b = 9.4, J3,10a = 5.4 Hz, 1H, H-10 aN), 3.34 (s, 3H, H-11N), 3.29 (s, 3H, H-11X), 3.22 (t, J3,10b = J10a,10b = 9.4 Hz, 1H, H-10bN), 3.14 (dd, J10a,10b = 9.5, J3,10a = 5.7 Hz, 1H, H-10aX), 2.93 (br s, 2H, H-4X and H-1N), 2.84 (t, J3,10b = J10a,10b = 9.5 Hz, 1H, H-10bX), 2.79 (br s, 1H, H-1X), 2.75 (br s, 1H, H-4N), 2.42 (m, 1H, H-3X), 1.81 (dt, J3,10b = 9.4, J3,10a = J2,3 = 5.3 Hz, 1H, H-3N), 1.33 (br s, 4H, H-7X and H-7N), 1.23 (s, 12H, H-9X), 1.17 (s, 12H, H-9N), 0.78 (dd, J2,3 = 5.2, J1,2 = 3.3 Hz, 1H, H-2N), 0.08 (br d, J2,3 = 5.9 Hz, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 138.4 (CH, C-6X), 136.9 (CH, C-5N), 135.9 (CH, C-6N), 131.5 (CH, C-5X), 83.0 (2C, C-8X), 82.9 (2C, C-8N), 77.5 (CH2, C-10N), 76.5 (CH2, C-10X), 58.6 (2CH3, C-11X and C-11N), 48.4 (CH2, C-7X), 46.7 (CH2, C-7N), 44.4 (CH, C-1X), 44.2 (CH, C-4X), 44.1 (CH, C-4N), 44.0 (CH, C-1N), 42.3 (CH, C-3N), 41.9 (CH, C-3X), 24.7 (4CH3, C-9X), 24.6 (4CH3, C-9N), C-2 signals missing. 11B NMR (96 MHz, CDCl3) δ 33 8.
(a) Procedure A: reaction time: 24 h. Yield: 29% (17.3 mg), endo/exo 35:65.
(b) Procedure B: reaction time: 24 h. Yield: 32% (19 mg), endo/exo 17:83. Reaction time: 72 h. Yield: 38% (22.5 mg), endo/exo 17:83.
Boronates 2e-X and 2e-N (yellowish oil). IR (film) νmax 2957, 2928, 2870, 1468, 1454, 1404, 1371, 1146, 853, 679 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.35–7.08 (m, 10H, ArH-X and ArH-N), 6.30 (dd, J5,6 = 5.6, J1,6 = 3.2 Hz, 1H, H-6X), 6.26 (dd, J5,6 = 5.6, J4,5 = 3.3 Hz, 1H, H-5N), 6.15 (dd, J5,6 = 5.6, J1,2 = 2.9 Hz, 1H, H-6N), 5.79 (dd, J5,6 = 5.6, J4,5 = 2.7 Hz, 1H, H-5X), 3.51 (dd, J2,3 = 5.8, J = 3.2 Hz, 1H, H-3X), 3.16 (br s, 1H, H-4X), 3.11 (br s, 1H, H-1N), 2.95 (br s, 2H, H-1X and H-4N), 2.86 (br d, J2,3 = 5.9 Hz, 1H, H-3N), 1.63 (br d, J7a,7b = 8.6, 1H, H-7aN), 1.56 (br d, J7a,7b = 8.3 Hz, 1H, H-7aX), 1.49–1.41 (m, 1H, H-7bN), 1.42–1.36 (m, 2H, H-7bX and H-2N), 1.25 (s, 12H, H-9X), 1.22 (s, 12H, H-9N), 1.02 (dd, J2,3 = 5.9, J = 1.8, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 146.7 (C, Ar-N), 145.1 (C, Ar-X), 138.6 (CH, C-6X), 137.6 (CH, C-5N), 136.5 (CH, C-6N), 132.1 (CH, C-5X), 128.2 (2CH, Ar-N), 128.1 (2CH, Ar-X), 127.7 (2CH, Ar-X), 127.5 (2CH, Ar-N), 125.5 (CH, Ar-X), 125.3 (CH, Ar-N), 83.2 (2C, C-8X), 83.1 (2C, C-8N), 49.0 (CH2, C-7X), 48.9 (CH, C-4X), 48.0 (CH2, C-7N), 47.9 (CH, C-4N), 46.5 (2CH, C-3X and C-3N), 45.9 (CH, C-1X), 45.2 (CH, C-1N), 24.9 (2CH3, C-9N), 24.8 (2CH3, C-9X), 24.7 (2CH3, C-9X), 24.6 (2CH3, C-9N), C-2 signals missing. 11B NMR (96 MHz, CDCl3) δ 33.3. HRMS (APCI) calcd for C19H26BO2 (M + H)+ 297.2020, found 297.2033.
(a) Procedure A: reaction time: 24 h. Yield: 20% (11.2 mg), endo/exo 40:60.
(b) Procedure B: reaction time: 24 h. Yield: 20% (11.2 mg), endo/exo 36:64. Reaction time: 72 h. Yield: 31% (17.4 mg), endo/exo 29:71.
Boronates 2f-X and 2f-N (yellowish oil). IR (film) νmax 3059, 2974, 2931, 2870, 1492, 1371, 1315, 1143, 1091, 1014, 972, 848, 798, 729 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.24 (s, 4H, H-11N and H-12N), 7.17 (br d, J11,12 = 8.5 Hz, 2H, H-11X), 7.07 (br d, J12,11 = 8.5 Hz, 2H, H-12X), 6.31 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6X), 6.25 (dd, J5,6 = 5.7, J4,5 = 3.1 Hz, 1H, H-5N), 6.15 (dd, J5,6 = 5.7, J1,6 = 2.9 Hz, 1H, H-6N), 5.76 (dd, J5,6 = 5.6, J4,5 = 2.8 Hz, 1H, H-5X), 3.45 (dd, J2,3 = 6.0, J3,4 = 3.8 Hz, 1H, H-3X), 3.11 (br s, 2H, H-4X and H-1N), 2.94 (br s, 2H, H-1X and H-4N), 2.80 (br d, J2,3 = 5.6 Hz, 1H, H-3N), 1.58–1.51 (m, 2H, H-7X and H-7N), 1.47 (br d, J7a,7b = 8.3, 1H, H-7N), 1.40 (m, 1H, H-7X), 1.33 (m, 1H, H-2N), 1.25 (s, 12H, H-9X), 1.22 (s, 12H, H-9N), 0.95 (dd, J2,3 = 6.0, J1,2 = 2.0 Hz, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 145.2 (C, C-10N), 143.6 (C, C-10X), 138.9 (CH, C-6X), 137.4 (CH, C-5N), 136.7 (CH, C-6N), 131.8 (CH, C-5X), 131.2 (C, C-13X), 129.4 (2CH, C-11X), 128.8 (2CH, C-11N), 128.2 (2CH, C-12N), 127.8 (2CH, C-12X), 83.3 (2C, C-8X), 83.2 (2C, C-8N), 49.1 (CH2, C-7X), 48.9 (CH, C-4X), 47.9 (CH, C-4N and CH2, C-7N), 46.1 (2CH, C-3N and C-3X), 45.8 (CH, C-1X), 45.1 (CH, C-1N), 24.9 (2CH3, C-9N), 24.8 (2CH3, C-9X), 24.7 (2CH3, C-9X), 24.6 (2CH3, C-9N), C-2 and C-13N signals missing. 11B NMR (96 MHz, CDCl3) δ 33.3. HRMS (APCI) calcd for C19H25BClO2 (M + H)+ 331.1631, found 331.1628.
(a) Procedure A: reaction time: 24 h. Yield: 19% (18.7 mg), endo/exo 37:63.
(b) Procedure B: reaction time: 24 h. Yield: 24% (23.6 mg), endo/exo 10:90. Reaction time: 72 h. Yield: 45% (44.3 mg), endo/exo 10:90.
Boronates 2g-X and 2g-N (yellowish oil). IR (film) νmax 3045, 2926, 1715, 1445, 1354, 1265, 1080, 737, 664, 600 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.78–7.26 (m, 8H, ArH-X and ArH-N) 6.34 (dd, J5,6 = 5.6, J1,6 = 2.9 Hz, 1H, H-6X), 6.27 (dd, J5,6 = 5.5, J4,5 = 3.0 Hz, 1H, H-5N), 6.17 (dd, J5,6 = 5.5, J1.6 = 2.8 Hz, 1H, H-6N), 5.76 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 1H, H-5X), 3.53 (dd, J2,3 = 5.8, J3,4 = 3.3 Hz, 1H, H-3X), 3.15 (br s, 2H, H-4X and H-1N), 2.98 (br s, 2H, H-1X and H-4N), 2.89 (br d, J2,3 = 5.7 Hz, 1H, H-3N), 1.60–1.45 (m, 3H, H-7aX, and H-7N), 1.44–1.34 (m, 2H, H-7bX and H-2N), 1.26 (s, 12H, H-9X), 1.23 (s, 12H, H-9N), 1.02 (dd, J2,3 = 5.8, J1,2 = 2.1 Hz, 1H, H-2X). 13C NMR (75 MHz, CDCl3) δ 147.7 (C, Ar-N), 146.1 (C, Ar-X), 139.1 (CH, C-6X), 137.4 (CH, C-5N), 136.7 (CH, C-6N), 131.7 (CH, C-5X), 131.4 (CH, Ar-X), 130.6 (CH, Ar-N), 130.4 (C, JC,F = 29.5 Hz, Ar-X), 128.6 (CH, Ar-N), 128.1 (CH, Ar-X), 125.0 (CH, Ar-N), 124.8 (CH, JC,F = 3.7 Hz, Ar-X), 122.6 (CH, Ar-N), 122.5 (CH, Ar-N), 122.4 (CH, JC,F = 3.7 Hz, Ar-X), 83.3 (2C, C-8X), 83.2 (2C, C-8N), 49.2 (CH2, C-7X), 48.9 (CH, C-4X), 48.0 (CH2, C-7N), 47.5 (CH, C-4N), 46.5 (2CH, C-3X and C-3N), 45.9 (CH, C-1X), 45.2 (CH, C-1N), 24.9 (2CH3, C-9N), 24.8 (2CH3, C-9X), 24.7 (2CH3, C-9X), 24.6 (2CH3, C-9N), C-12N, C-2 and CF3 signals missing. 11B NMR (96 MHz, CDCl3) δ 33.6. 19F NMR (282 MHz, CDCl3) δ −62.5. HRMS (APCI) calcd for C20H25BF3O2 (M + H)+ 365.1894, found 365.1879.
(a) Procedure A: reaction conditions: 12 h at 170 °C. Yield: 21% (10.6 mg), endo/exo 41:59. Reaction conditions: 12 h at 80 °C. Yield: 91% (45.8 mg), endo/exo 5:95.
(b) Procedure B: reaction time: 12 h. Yield: 83% (41.8 mg), endo/exo 6:94.
Boronates 2h-X and 2h-N (white solid, mp 81.5–83.3 °C). IR (KBr) νmax 3065, 2976, 2864, 1371, 1327, 1314, 1215, 1138, 1051 856, 698, 611 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.39–7.00 (m, 10H, ArH-X and ArH-N), 6.25 (dd, J5,6 = 5.4, J1,6 = 2.9 Hz, 1H, H-6N), 6.21 (dd, J5,6 = 5.4, J4,5 = 3.0 Hz, 1H, H-5N), 6.03 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 1H, H-5X), 5.90 (dd, J5,6 = 5.6, J1,6 = 2.6 Hz, 1H, H-6X), 3.58 (br s, 1H, H-1X), 3.48 (br s, 1H, H-1N), 2.88 (br s, 2H, H-4X and H-4N), 2.48 (dd, J3n,3x = 11.5, J3x,4 = 3.9 Hz, 1H, H-3xX), 2.05 (br d, J3n,3x = 11.2 Hz, 1H, H-3nN), 1.93 (dd, J3n,3x = 11.2, J3x,4 = 3.4 Hz, 1H, H-3xN), 1.51–1.39 (m, 4H, H-3nX, H-7aX, and H-7N), 1.31 (br d, J7a,7b = 8.3 Hz, 1H, H-7bX), 1.11 (s, 6H, H-9X), 1.10 (s, 12H, H-9X and H-9N), 1.08 (s, 6H, H-9N). 13C NMR (75 MHz, CDCl3) δ 146.4 (C, Ar-X), 138.8 (CH, C-5N), 136.4 (CH, C-6X), 136.0 (CH, C-6N), 134.9 (CH, C-5X), 128.1 (2CH, Ar-X), 127.8 (2CH, Ar-X), 127.6 (2CH, Ar-N), 125.2 (2CH, Ar-N), 124.5 (CH, Ar-X), 83.3 (2C, C-8X), 83.2 (2C, C-8N), 49.0 (CH2, C-7X), 47.8 (CH, C-1X), 47.3 (CH, C-1N), 47.2 (CH2, C-7N), 43.4 (CH, C-4N), 42.4 (CH, C-4X), 39.0 (CH2, C-3N) 35.6 (CH2, C-3X), 24.5 (2CH3, C-9N) 24.3 (2CH3, C-9X), 24.2 (4CH3, C-9X and C-9X), C-2, C-10N and C-13N signals missing. 11B NMR (96 MHz, CDCl3) δ 33.3. HRMS (APCI) calcd for C19H26BO2 (M + H)+ 297.1010, found 297.2016.
(a) Procedure A: reaction time: 24 h. Yield: 72% (84.3 mg), endo/exo 9:91.
(b) Procedure B: reaction time: 24 h. Yield: 15% (17.6 mg), endo/exo 9:91.
Boronate 2i-X (major compound, yellowish liquid). IR (film) νmax 3055, 2958, 2927, 2866, 1456, 1371, 1354, 1303, 1145, 719 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.12 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6), 6.00 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 1H, H-5), 2.75 (br s, 2H, H-1 and H-4), 2.03 (dd, J3n,3x = 11.3, J = 3.8 Hz, 1H, H-3x), 1.28–1.12 (m, 2H, H-7), 1.24 (s, 12H, H-9), 0.81 (s, 3H, H-10), 0.53 (dd, J3n,3x = 11.3, J = 2.5 Hz, 1H, H-3n). 13C NMR (75 MHz, CDCl3) δ 136.3 (CH, C-6), 133.8 (CH, C-5), 83.0 (2C, C-8), 49.7 (CH2, C-7), 48.9 (CH, C-4), 43.4 (CH, C-1), 36.6 (CH2, C-3), 24.6 (4CH3, C-9), 22.1 (CH3, C-10), C-2 signal missing. 11B NMR (96 MHz, CDCl3) δ 35.0.
Boronates 2i-X and 2i-N (yellow liquid). IR (film) νmax 2954, 2924, 2852, 1604, 1463, 1446, 1435, 1359, 1303, 1145, 1022, 746 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.14 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6N), 6.12 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6X), 6.00 (dd, J5,6 = 5.6, J4,5 = 2.9 Hz, 2H, H-5X and H-5N), 2.76 (br s, 3H, H-1X, H-4X and H-4N), 2.53 (br s, 1H, H-1N), 2.03 (dd, J3n,3x = 11.3, J = 3.8 Hz, 1H, H-3xX), 1.52–1.12 (m, 6H, H-7X, H-3N and H-7N), 1.24 (s, 12H, H-9X), 1.19 (s, 12H, H-9N), 1.13 (s, 3H, H-10N), 0.81 (s, 3H, H-10X), 0.53 (dd, J3n,3x = 11.3, J = 2.5 Hz, 1H, H-3nX). 13C NMR (75 MHz, CDCl3) δ 137.0 (CH, C-6N), 136.6 (CH, C-5N), 136.3 (CH, C-6X), 133.8 (CH, C-5X), 83.0 (2C, C-8X), 82.8 (2C, C-8N), 50.0 (CH, C-1N), 49.7 (CH2, C-7X), 48.9 (CH, C-4X), 45.6 (CH2, C-3N), 43.4 (CH, C-1X), 42.9 (CH, C-4N), 37.9 (CH2, C-7N), 36.6 (CH2, C-3X), 24.6 (8CH3, C-9X and C-9N), 24.2 (CH3, C-10N), 22.1 (CH3, C-10X), C-2 signals missing. 11B NMR (96 MHz, CDCl3) δ 34.3. HRMS (APCI) calcd for C14H24BO2 (M + H)+ 235.1864, found 235.1770.
Tandem Diels–Alder reaction of alkenylboronates – oxidation: synthesis of alcohols
:61.:85.
Alcohols 3b-X and 3b-N (yellowish oil). IR (film) νmax 3404, 2957, 2922, 2851, 2358, 1717, 1024, 849, 667 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.48 (dd, J5,6 = 5.6, J4,5 = 3.1 Hz, 1H, H-5N), 6.11 (dd, J5,6 = 5.7, J4,5 = 2.6 Hz, 1H, H-5X), 6.09 (m, 1H, H-6N), 6.02 (dd, J5,6 = 5.7, J1,6 = 3.1 Hz, 1H, H-6X), 3.91 (br s, 1H, H-2N), 3.33 (br s, 1H, H-2X), 2.89 (br s, 1H, H-1N), 2.67 (br s, 1H, H-1X), 2.65 (br s, 1H, H-4X), 2.50 (br s, 1H, H-4N), 1.79 (d, J7a,7b = 8.5, 1H, H-7aX), 1.66–1.56 (m, 2H, H-3X and H-7bX), 1.54–1.07 (m, 10H, H-8X, H-9X, H-7N, H-8N and H-9N), 1.02–0.98 (m, 1H, H-3N), 0.93 (t, J9,10 = 7.2 Hz, 3H, H-10N), 0.90 (t, J9,10 = 6.9 Hz, 3H, H-10X). 13C NMR (75 MHz, CDCl3) δ 141.2 (CH, C-5N), 137.3 (CH, C-5X), 133.8 (CH, C-6X), 131.6 (CH, C-6N), 79.9 (CH, C-2N), 79.0 (CH, C-2X), 50.9 (CH, C-4X), 50.8 (CH, C-3N), 50.5 (CH, C-3X), 48.3 (CH, C-1N), 47.3 (CH, C-4N), 46.6 (CH2, C-7X), 45.2 (CH2, C-7N), 44.5 (CH, C-1X), 36.9 (CH2, C-8N), 35.8 (CH2, C-8X), 21.7 (CH2, C-9N), 21.6 (CH2, C-9X), 14.2 (2CH3, C-10X and C-10N). HRMS (APCI) calcd for C10H17O (M + H)+ 153.1274, found 153.1277.
:86.
Alcohol 3c-X (major compound, yellowish oil). IR (film) νmax 3362, 2963, 2868, 2359, 2344, 2322, 1558, 1541, 1489, 1456, 1373, 1339, 1214, 995, 849, 718 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.13 (dd, J5,6 = 5.7, J1,6 = 2.7 Hz, 1H, H-5), 6.05 (dd, J5,6 = 5.7, J4,5 = 3.1 Hz, 1H, H-6), 3.54 (t, J9,10 = 6.3 Hz, 2H, H-10), 3.36 (br s, 1H, H-2), 2.68 (br s, 1H, H-1), 2.66 (br s, 1H, H-4), 1.89–1.77 (m, 3H, H-7a and H-9), 1.68–1.59 (m, 2H, H-3 and H-7b), 1.52 (br s, 1H, OH), 1.47–1.28 (m, 2H, H-8). 13C NMR (75 MHz, CDCl3) δ 137.0 (CH, C-5), 134.2 (CH, C-6), 78.8 (CH, C-2), 51.0 (CH, C-4), 49.8 (CH, C-3), 46.6 (CH2, C-7), 45.1 (CH2, C-10), 44.5 (CH, C-1), 31.5 (CH2, C-9), 30.7 (CH2, C-8).
Alcohols 3c-X and 3c-N (yellowish oil). IR (film) νmax 3345, 3327, 3059, 2964, 2935, 2870, 1456, 1339, 1028, 849, 717, 648 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.49 (dd, J5,6 = 5.8, J4,5 = 3.0 Hz, 1H, H-5N), 6.13 (dd, J5,6 = 5.7, J4,5 = 2.7 Hz, 1H, H-5X), 6.11 (m, 1H, H-6N), 6.05 (dd, J5,6 = 5.7, J1,6 = 3.1 Hz, 1H, H-6X), 3.93 (br s, 1H, H-2N), 3.58 (t, J9,10 = 6.5 Hz, 2H, H-10N), 3.54 (t, J9,10 = 6.3 Hz, 2H, H-10X), 3.36 (br s, 1H, H-2X), 2.91 (br s, 1H, H-1N), 2.68 (br s, 1H, H-1X), 2.66 (br s, 1H, H-4X), 2.51 (br s, 1H, H-4N), 1.98–1.77 (m, 7H, H-7aX, H-9X, H-8N and H-9N), 1.68–1.49 (m, 5H, H-3X, H-7bX, OH-X and H-7N), 1.47–1.28 (m, 2H, H-8X), 1.04–0.96 (m, 1H, H-3N). 13C NMR (75 MHz, CDCl3) δ 141.1 (CH, C-5N), 137.0 (CH, C-5X), 134.2 (CH, C-6X), 131.8 (CH, C-6N); 79.6 (CH, C-2N), 78.8 (CH, C-2X), 51.0 (CH, C-4X), 50.2 (CH, C-3N), 49.8 (CH, C-3X), 48.3 (CH, C-1N), 47.4 (CH, C-4N), 46.6 (CH2, C-7X), 45.2 (CH2, C-7N), 45.1 (2CH2, C-10X and C-10N), 44.5 (CH, C-1X), 31.8 (CH2, C-8N), 31.5 (2CH2, C-9X and C-9N), 30.7 (CH2, C-8X). HRMS (APCI) calcd for C10H15ClO (M + H–H2O)+ 169.0779, found 169.0810.
:88.
Alcohol 3d-X (major compound, yellowish oil). IR (film) νmax 3400, 2970, 2920, 2891, 2872, 2850, 1109, 1033, 717 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.11 (dd, J5,6 = 5.7, J1,6 = 2.7 Hz, 1H, H-5), 6.06 (dd, J5,6 = 5.5, J4,5 = 3.3 Hz, 1H, H-6), 3.43 (br s, 1H, H-2), 3.33 (s, 3H, H-9), 3.22 (dd, Jgem = 14.4, J3,8 = 8.0 Hz, 1H, H-8), 3.19 (dd, Jgem = 14.4, J3,8 = 7.9 Hz, 1H, H-8), 2.78 (br s, 1H, H-4), 2.71 (br s, 1H, H-1), 1.90 (m, 1H, H-3), 1.85 (br d, J7a,7b = 8.5 Hz, 1H, H-7b), 1.75 (br s, 1H, OH), 1.66 (dd, J7a,7b = 8.5, J2,7a = 1.6 Hz, 1H, H-7a). 13C NMR (75 MHz, CDCl3) δ 137.1 (CH, C-5), 134.4 (CH, C-6), 76.2 (CH, C-2), 75.8 (CH2, C-8), 58.8 (CH3, C-9), 50.7 (CH, C-3), 50.5 (CH, C-1), 46.7 (CH2, C-7), 43.1 (CH, C-4).
Alcohols 3d-X and 3d-N (yellowish oil). IR (film) νmax 3415, 3059, 2970, 2922, 2872, 2827, 1134, 1111, 1083, 1035, 985, 918, 717 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.47 (dd, J5,6 = 5.7, J1,6 = 3.3 Hz, 1H, H-5N), 6.13 (dd, J5,6 = 5.7, J4,5 = 2.9 Hz, 1H, H-6N), 6.11 (dd, J5,6 = 5.7, J1,6 = 2.7 Hz, 1H, H-5X), 6.06 (dd, J5,6 = 5.5, J4,5 = 3.3 Hz, 1H, H-6X), 4.00 (br s, 1H, H-2N), 3.56–3.49 (m, 1H, H-8N), 3.43 (br s, 1H, H-2X), 3.36 (s, 3H, H-9N), 3.40–3.33 (m, 1H, H-8N), 3.33 (s, 3H, H-9X), 3.21 (dd, Jgem = 17.0, J3,8 = 8.0 Hz, 1H, H-8X), 3.18 (dd, Jgem = 17.1, J3,8 = 7.9 Hz, 1H, H-8X), 2.93 (br s, 1H, H-1N), 2.78 (br s, 1H, H-4X), 2.71 (br s, 1H, H-1X), 2.65 (br s, 1H, H-4N), 1.9 (m, 1H, H-3X), 1.85 (br d, J7a,7b = 8.5 Hz, 1H, H-7X), 1.76 (br s, 1H, OH-X), 1.66 (dd, J7a,7b = 8.5, J3,7a = 1.6 Hz, 1H, H-7X), 1.48 (m, 2H, H-7N), 1.43–1.29 (m, 1H, H-3N). 13C NMR (75 MHz, CDCl3) δ 140.5 (CH, C-5N), 137.1 (CH, C-5X), 134.4 (CH, C-6X), 132.4 (CH, C-6N), 76.7 (CH, C-2N), 76.2 (CH, C-2X), 75.8 (CH2, C-8X), 75.7 (CH2, C-8N), 58.9 (CH3, C-9N), 58.8 (CH3, C-9X), 50.8 (CH, C-3N), 50.7 (CH, C-3X), 50.5 (CH, C-1X), 48.0 (CH, C-1N), 46.7 (CH2, C-7X), 45.2 (CH2, C-7N), 44.9 (CH, C-4N), 43.1 (CH, C-4X). HRMS (APCI) calcd for C9H13O (M + H–H2O)+ 137.0961, found 137.0938.
:87.
Alcohols 3e-X and 3e-N (yellowish oil). IR (film) νmax 3061, 3323, 2968, 2939, 2922, 1033, 746, 717, 698 cm−1. 1H NMR (300 MHz) δ 7.39–7.14 (m, 10H, ArH-X and ArH-N), 6.64 (dd, J5,6 = 5.7, J4,5 = 3.2 Hz, 1H, H-5N), 6.25 (dd, J5,6 = 5.7, J1,6 = 2.9 Hz, H-6N), 6.20 (dd, J5,6 = 5.7, J5,4 = 3.3 Hz, 1H, H-5X), 6.07 (br d, J5,6 = 5.7 Hz, 1H, H-6X), 4.42 (br s, 1H, H-2N), 4.04 (br s, 1H, H-2X), 3.02 (br s, 4H, H-3X, H-4X, H-1N and H-4N), 2.83 (br s, 1H, H-1X), 2.36 (t, J2,3 = J3,4 = 3.0 Hz, 1H, H-3N), 2.05 (br d, J7a,7b = 8.3 Hz, 1H, H-7aX), 1.79 (br d, J7a,7b = 8.5 Hz, 1H, H-7aN), 1.76 (br d, J7a,7b = 8.3 Hz, 1H, H-7bX), 1.67–1.61 (m, 1H, H-7bN). 13C NMR (75 MHz) δ 143.8 (C, Ar-N), 143.3 (C, Ar-X), 141.3 (CH, C-5N), 137.6 (CH, C-6X), 134.0 (CH, C-5X), 132.9 (CH, C-6N), 128.5 (2CH, Ar-N), 128.0 (2CH, Ar-X), 127.8 (2CH, Ar-X), 127.2 (2CH, Ar-N), 126.0 (2CH, Ar-X and Ar-N), 80.7 (CH, C-2N), 79.4 (CH, C-2X), 55.4 (CH, C-3X), 55.3 (CH, C-3N), 51.3 (CH, C-1X), 48.6 (CH, C-1N), 48.1 (CH, C-4N), 47.3 (CH2, C-7X), 47.1 (CH, C-4X), 45.7 (CH2, C-7N). HRMS (APCI) calcd for C13H13 (M + H–H2O)+ 169.1012, found 169.1041.
:87.
Alcohols 3f-X and 3f-N (yellowish oil). IR (film) νmax 3361, 3340, 2964, 2916, 2848, 1490, 1091, 1033, 1012, 798, 727 cm−1. 1H NMR (300 MHz) δ 7.31–7.19 (m, 6H, ArH-X and ArH-N), 7.14–7.09 (m, 2H, ArH-X), 6.63 (dd, 1H, J5,6 = 5.9, J4,5 = 3.3 Hz, H-5N), 6.25 (dd, 1H, J5,6 = 5.8, J1,6 = 3.0 Hz, H-6N), 6.16 (dd, 1H, J5,6 = 5.7, J4,5 = 3.3 Hz, H-5X), 6.04 (br d, 1H, J5,6 = 5.70 Hz, H-6X), 4.35 (br s, 1H, H-2N), 3.98 (br s, 1H, H-2X), 3.03 (br s, 1H, H-4N), 2.99 (br s, 3H, H-3X, H-4X and H-1N), 2.83 (br s, 1H, H-1X), 2.32 (t, 1H, J2,3 = J3,4 = 3.0 Hz, H-3N), 2.04 (br d, 1H, J7a,7b = 8.6 Hz, H-7aX), 1.76 (br d, J7a,7b = 8.6 Hz, 1H, H-7bX), 1.76–1.60 (m, 2H, H-7N). 13C NMR (75 MHz, CDCl3) δ 141.8 (2C, Ar-X and Ar-N), 141.2 (CH, C-5N), 137.3 (CH, C-6X), 134.2 (CH, C-5X), 132.9 (CH, C-6N), 131.8 (C, Ar-X), 129.1 (2CH, Ar-X), 128.5 (4CH, Ar-N), 128.1 (2CH, Ar-X), 80.9 (C, C-2N), 79.5 (C, C-2X), 54.8 (CH, C-3N), 54.6 (CH, C-3X), 51.3 (CH, C-1X), 48.6 (CH, C-4N), 47.9 (CH, C-1N), 47.3 (CH2, C-7X), 47.0 (CH, C-4X), 45.7 (CH2, C-7N), C-8N not detected. HRMS (APCI) calcd for C13H12Cl (M + H–H2O)+203.0628, found 203.0586.
:91.
Alcohols 3g-X and 3g-N (yellowish oil). IR (film) νmax 3343, 3308, 2970, 2916, 2359, 2344, 1331, 1165, 1124, 1074, 1034, 795, 721, 669 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.71–7.31 (m, 8H, ArH-X and ArH-N), 6.65 (dd, J5,6 = 5.4, J4,5 = 3.1 Hz, 1H, H-5N), 6.27 (dd, J5,6 = 5.4, J1,6 = 2.8 Hz, 1H, H-6N), 6.20 (dd, J5,6 = 5.7, J1,6 = 3.3 Hz, 1H, H-6X), 6.05 (dd, J5,6 = 5.7, J4,5 = 2.7 Hz, 1H, H-5X), 4.39 (m, 1H, H-2N), 4.03 (br s, 1H, H-2X), 3.07 (m, 2H, H-3X and H1–N), 3.04 (br s, 2H, H-4X and H-4N), 2.85 (br s, 1H, H-1X), 2.10 (m, 1H, H-3N), 2.06 (br d, J7a,7b = 8.8 Hz, 1H, H-7aX), 1.90 (br s 1H, OH-X), 1.82–1.76 (m, 1H, H-7bX), 1.76–1.63 (m, 2H, H-7N). 13C NMR (75 MHz, CDCl3) δ 144.5 (C, Ar-N), 144.3 (C, Ar-X), 141.0 (CH, C-5N), 137.2 (CH, C-5X), 134.4 (CH, C-6X), 133.1 (CH, C-6N), 131.2 (CH, Ar-X), 130.3 (C, JC,F = 32.0 Hz, Ar-X), 129.7 (CH, Ar-N), 128.9 (CH, Ar-N), 128.4 (CH, Ar-X), 126.1 (CH, Ar-N), 124.6 (CH, Ar-N), 124.5 (CH, JC,F = 3.5 Hz, Ar-X), 122.9 (CH, JC,F = 3.9 Hz, ArX), 122.4 (CH, Ar-N), 80.8 (CH, C-2N), 79.4 (CH, C-2X), 55.1 (CH, C-3N), 55.0 (CH, C-3X), 51.4 (CH, C-1X), 48.6 (CH, C-4N), 47.3 (CH2, C-7X), 47.8 (CH, C-1N), 47.0 (CH, C-4X), 45.7 (CH, C-7N). 19F NMR (282 MHz, CDCl3) δ −62.6. HRMS (APCI) calcd for C14H12F3 (M + H – H2O)+ 237.0886, found 237.0902.
:94.
Alcohol 3h-X (major compound, white solid, mp 62.5–63.0 °C). IR (KBr) νmax 3364, 2986, 2970, 2945, 1493, 1447, 1274, 1061, 1028, 989, 894, 758, 721, 698 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.43–7.19 (m, 5H, ArH), 6.17 (dd, J5,6 = 5.6, J4,5 = 3.0 Hz, 1H, H-5), 5.78 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6), 3.08–3.03 (m, 1H, H-1), 2.98 (br s, 1H, H-4), 2.16 (br d, J7a,7b = 8.6 Hz, 1H, H-7b), 2.13 (dd, J3n,3x = 12.2, J3n,4 = 2.3 Hz, 1H, H-3n), 2.03 (dd, J3n,3x = 12.2, J3x,4 = 3.5 Hz, 1H, H-3x), 1.95 (br s, 1H, OH), 1.75–1.68 (m, 1H, H-7a). 13C NMR (75 MHz, CDCl3) δ 146.6 (C, Ar), 138.9 (CH, C-5), 134.5 (CH, C-6), 128.1 (2CH, Ar), 127.0 (3CH, Ar), 82.7 (C, C-2), 54.3 (CH, C-1), 48.2 (CH2, C-7), 43.1 (CH2, C-3), 41.9 (CH, C-4).
Alcohols 3h-X and 3h-N (white solid). IR (KBr) νmax 3366, 2970, 2945, 1491, 1447, 1274, 1061, 1028, 989, 895, 758, 721, 698 cm−1. 1H NMR (300 MHz, CDCl3) δ 7.63–7.19 (m, 10H, ArH-X and ArH-N), 6.58 (dd, J5,6 = 5.7, J1,6 = 3.0 Hz, 1H, H-6N), 6.33 (dd, J5,6 = 5.7, J4,5 = 3.0 Hz, 1H, H-5N), 6.17 (dd, J5,6 = 5.6, J4,5 = 3.0 Hz, 1H, H-5X), 5.78 (dd, J5,6 = 5.6, J1,6 = 3.1 Hz, 1H, H-6X), 3.24–3.20 (m, 1H, H-4N), 3.08–3.03 (m, 1H, H-1X), 2.98 (br s, 2H, H-4X and H-1N), 2.49 (dd, J3x,3n = 12.6, J3x,4 = 3.7 Hz, 1H, H-3xN), 2.16 (br d, J7a,7b = 8.6 Hz, 1H, H-7bX), 2.13 (dd, J3n,3x = 12.2, J3n,4 = 2.3 Hz, 1H, H-3nX), 2.03 (dd, J3n,3x = 12.2, J3x,4 = 3.5 Hz, 1H, H-3xX), 1.95 (br s, 1H, OH-X), 1.83 (br s, 1H, OH–N), 1.75–1.68 (m, 1H, H-7aX), 1.63–1.60 (m, 2H, H-7N), 1.52–1.44 (m, 1H, H-3nN). 13C NMR (75 MHz, CDCl3) δ 146.6 (2C, Ar-X and Ar-8N), 141.3 (C, C-6N), 138.9 (CH, C-5X), 134.5 (CH, C-6X), 133.6 (C, C-5N), 129.2 (CH, Ar-N), 128.1 (4CH, Ar-X and Ar-N), 127.0 (3CH, Ar-X), 82.7 (C, C-2X), 54.3 (CH, C-1X), 53.2 (CH, C-4N), 49.3 (CH2, C-7N), 48.2 (CH2, C-7X), 44.8 (CH2, C-3N), 43.3 (CH2, C-1N), 43.1 (CH2, C-3X), 41.9 (CH, C-4X). C-2N signal missing. HRMS (ESI) calcd for C13H14ONa (M + Na)+ 209.1250, found 209.0937.
:91.
Alcohols 3i-X and 3i-N (yellowish liquid). IR (film) νmax 3381, 3061, 2956, 2924, 2868, 2852, 1446, 1330, 1251, 1109, 939, 887, 729, 705 cm−1. 1H NMR (300 MHz, CDCl3) δ 6.42 (dd, J5,6 = 5.63, J1,6 = 3.1 Hz, 1H, H-6N), 6.19 (dd, J5,6 = 5.6, J4,5 = 3.0 Hz, 1H, H-5N), 6.12 (dd, J5,6 = 5.5, J4,5 = 2.9 Hz, 1H, H-5X), 6.06 (dd, J5,6 = 5.5, J1,6 = 3.2 Hz, 1H, H-6X), 2.82 (br s, 2H, H-4X and H-4N), 2.65 (br s, 1H, H-1N), 2.48 (br s, 1H, H-1X), 1.92 (br d, J7a,7b = 8.5 Hz, 1H, H-7X), 1.80 (dd, J3n,3x = 12.2, J = 3.6 Hz, 1H, H-3xN), 1.68 (dd, J3n,3x = 12.1, J = 3.8 Hz, 1H, H-3xX), 1.59–1.48 (m, 2H, H-7N), 1.56 (br d, J7a,7b = 8.5 Hz, 1H, H-7X), 1.49 (s, 3H, H-8N), 1.28–1.21 (m, 1H, H-3nX), 1.22 (s, 3H, H-8X), 1.16 (dd, J3n,3x = 12.3, J = 3.3 Hz, 1H, H-3nN). 13C NMR (75 MHz, CDCl3) δ 140.0 (CH, C-6N), 138.4 (CH, C-5X), 134.5 (CH, C-6X), 133.5 (CH, C-5N), 79.1 (C, C-2X), 78.5 (C, C-2N), 54.9 (CH, C-1X), 53.8 (CH, C-1N), 49.5 (CH2, C-7N), 48.4 (CH2, C-7X), 44.9 (CH2, C-3N), 43.5 (CH2, C-3X), 43.0 (CH, C-4N), 42.3 (CH, C-4X), 28.2 (CH3, C-8N), 27.7 (CH3, C-8X). HRMS (APCI) calcd for C8H13O (M + H)+ 125.0966, found 125.0961.
Acknowledgements
We thank CONICET, Universidad Nacional de Rosario, Universidad Nacional del Nordeste, ANPCyT and Fundación Josefina Prats for financial support.Notes and references
- D. G. Hall, Boronic acids: preparation and applications in organic synthesis and medicine, Wiley-VCH, Weinheim, 2005 Search PubMed.
- G. Hilt and P. Bolze, Synthesis, 2005, 2091–2115 CrossRef CAS PubMed.
- J. E. Moore, M. York and J. P. A. Harrity, Synlett, 2005, 0860–0862 CAS.
- M. D. Helm, J. E. Moore, A. Plant and J. P. A. Harrity, Angew. Chem., Int. Ed., 2005, 44, 3889–3892 CrossRef CAS PubMed.
- P. M. Delaney, J. E. Moore and J. P. A. Harrity, Chem. Commun., 2006, 3323–3325 RSC.
- M. D. Helm, A. Plant and J. P. A. Harrity, Org. Biomol. Chem., 2006, 4, 4278–4280 CAS.
- E. Gomez-Bengoa, M. D. Helm, A. Plant and J. P. A. Harrity, J. Am. Chem. Soc., 2007, 129, 2691–2699 CrossRef CAS PubMed.
- M. D. Helm, A. Plant and J. P. A. Harrity, Synlett, 2007, 2885–2887 CAS.
- D. L. Browne, M. D. Helm, A. Plant and J. P. A. Harrity, Angew. Chem., Int. Ed., 2007, 46, 8656–8658 CrossRef CAS PubMed.
- P. M. Delaney, J. Huang, S. J. F. Macdonald and J. P. A. Harrity, Org. Lett., 2008, 10, 781–783 CrossRef CAS PubMed.
- P. M. Delaney, D. L. Browne, H. Adams, A. Plant and J. P. A. Harrity, Tetrahedron, 2008, 64, 866–873 CrossRef CAS PubMed.
- D. L. Browne, J. F. Vivat, A. Plant, E. Gomez-Bengoa and J. P. A. Harrity, J. Am. Chem. Soc., 2009, 131, 7762–7769 CrossRef CAS PubMed.
- R. S. Foster, J. Huang, J. F. Vivat, D. L. Browne and J. P. A. Harrity, Org. Biomol. Chem., 2009, 7, 4052–4056 CAS.
- D. L. Browne and J. P. A. Harrity, Tetrahedron, 2010, 66, 553–568 CrossRef CAS PubMed.
- J. D. Kirkham, P. M. Delaney, G. J. Ellames, E. C. Row and J. P. A. Harrity, Chem. Commun., 2010, 46, 5154–5156 RSC.
- R. S. Foster, H. Jakobi and J. P. A. Harrity, Tetrahedron Lett., 2011, 52, 1506–1508 CrossRef CAS PubMed.
- J. Kirkham, A. Leach, E. Row and J. P. A. Harrity, Synthesis, 2012, 44, 1964–1973 CrossRef CAS PubMed.
- J. D. Kirkham, S. J. Edeson, S. Stokes and J. P. A. Harrity, Org. Lett., 2012, 14, 5354–5357 CrossRef CAS PubMed.
- J. D. Kirkham, R. J. Butlin and J. P. A. Harrity, Angew. Chem., Int. Ed., 2012, 51, 6402–6405 CrossRef CAS PubMed.
- R. S. Foster, H. Jakobi and J. P. A. Harrity, Org. Lett., 2012, 14, 4858–4861 CrossRef CAS PubMed.
- D. S. Matteson and J. O. Waldbillig, J. Org. Chem., 1963, 28, 366–369 CrossRef CAS.
- I. S. Bengelsdorf, K. Kiyoshi, G. W. Willcockson and W. G. Woods, US Pat., 3135781, 1964.
- W. G. Woods and I. S. Bengelsdorf, J. Org. Chem., 1966, 31, 2769–2772 CrossRef CAS.
- D. S. Matteson and M. L. Talbot, J. Am. Chem. Soc., 1967, 89, 1123–1126 CrossRef CAS.
- D. A. Evans, W. L. Scott and L. K. Truesdale, Tetrahedron Lett., 1972, 13, 121–124 CrossRef.
- P. Martinez-Fresneda and M. Vaultier, Tetrahedron Lett., 1989, 30, 2929–2932 CrossRef CAS.
- K. Narasaka and I. Yamamoto, Tetrahedron, 1992, 48, 5743–5754 CrossRef CAS.
- C. Rasset and M. Vaultier, Tetrahedron, 1994, 50, 3397–3406 CrossRef CAS.
- J. D. Bonk and M. A. Avery, Tetrahedron: Asymmetry, 1997, 8, 1149–1152 CrossRef CAS.
- G. Lorvelec and M. Vaultier, Tetrahedron Lett., 1998, 39, 5185–5188 CrossRef CAS.
- R. A. Batey, A. N. Thadani and A. J. Lough, J. Am. Chem. Soc., 1999, 121, 450–451 CrossRef CAS.
- A. M. Sarotti, P. L. Pisano and S. C. Pellegrinet, Org. Biomol. Chem., 2010, 8, 5069–5073 CAS.
- E. Plettner, A. Mohle, M. T. Mwangi, J. Griscti, B. O. Patrick, R. Nair, R. J. Batchelor and F. Einstein, Tetrahedron: Asymmetry, 2005, 16, 2754–2763 CrossRef CAS PubMed.
- H. Yamamoto and K. Ishihara, Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- D. G. Hall, Boronic acids: preparation and applications in organic synthesis, medicine and materials, Wiley-VCH, Weinheim, 2nd completely rev. edn, 2011 Search PubMed.
- S. Roscales, Á. Rincón, E. Buxaderas and A. G. Csákÿ, Tetrahedron Lett., 2012, 53, 4721–4724 CrossRef CAS PubMed.
- S. Roscales and A. G. Csákÿ, Org. Lett., 2012, 14, 1187–1189 CrossRef CAS PubMed.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision D.01 Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- N. Grimblat and S. C. Pellegrinet, Org. Biomol. Chem., 2013, 11, 3733–3741 CAS.
- C. J. Collins and B. M. Benjamin, J. Am. Chem. Soc., 1967, 89, 1652–1661 CrossRef CAS.
- A. Padwa and W. Eisenberg, J. Am. Chem. Soc., 1972, 94, 5852–5858 CrossRef CAS.
- R. L. Snowden, Helv. Chim. Acta, 1983, 66, 1031–1038 CrossRef CAS PubMed.
- T. G. Waddell, A. D. Carter, T. J. Miller and R. M. Pagni, J. Org. Chem., 1992, 57, 381–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all novel compounds. Reaction coordinates and geometries of transition structures not included in the paper. See DOI: 10.1039/c4ra07415g |
| This journal is © The Royal Society of Chemistry 2014 |