
Synthesis and application of ultra-long Na0.44MnO2 submicron slabs as a cathode material for Na-ion batteries
Abstract
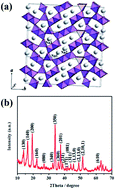
Novel ultra-long Na0.44MnO2 submicron slabs were fabricated through the sol–gel method, followed by high-temperature calcination. The material has a thickness ranging from 100 to 250 nm and a length varying from 10 μm to 40 μm. Electrochemical characterization indicates that the material can deliver a high capacity, larger than 120 mA h g−1 with stable cycling over 100 cycles in assembled non-aqueous Na-ion cells, the good performance of which is mainly attributed to the reduced sodium ion diffusion distance.
 Please wait while we load your content...
Please wait while we load your content...

