
Photo-responsive reversible micelles based on azobenzene-modified poly(carbonate)s via azide–alkyne click chemistry†
Abstract
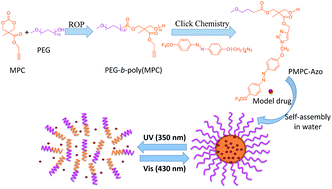
Photo-induced reversible amphipathic copolymer PMPC-azo was click conjugated by connecting amphiphilic poly(ethylene glycol)-modified poly(carbonate)s (PEG-b-poly(MPC)) and azide-functional trifluoromethoxy-azobenzene (azo-N3). The resulting copolymer self-assembled into spherical micelles with a hydrophobic azo core stabilized by a hydrophilic PEG corona in aqueous solution. As characterized by time-resolved UV-vis spectroscopy, dynamic light scattering (DLS) and transmission electron microscopy (TEM), these micelles showed reversible self-assembly and disassembly in aqueous solution under alternating UV and visible light irradiation. The model drug Nile Red (NR) was then successfully encapsulated into the micelles. Light-controlled release and re-encapsulation behaviors were demonstrated by fluorescence spectroscopy. The cell cytotoxicity of PMPC-azo micelles was also evidenced by MTT assay. This study provides a convenient method to construct smart nanocarriers for controlled release and re-encapsulation of hydrophobic drugs.
- This article is part of the themed collection: A Decade of Progress in Click Reactions Based on CuAAC
 Please wait while we load your content...
Please wait while we load your content...

