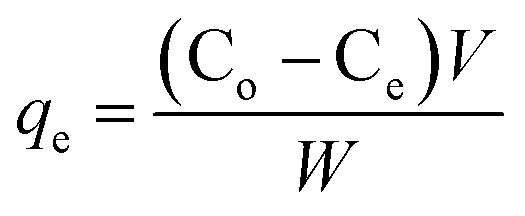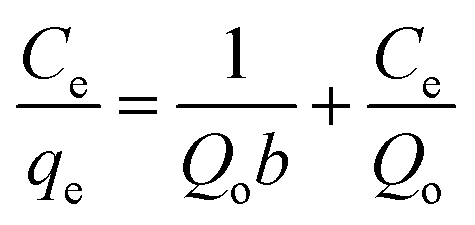DOI:
10.1039/C4RA07318E
(Paper)
RSC Adv., 2014,
4, 50510-50520
Facile in situ growth of Fe3O4 nanoparticles on hydroxyapatite nanorods for pH dependent adsorption and controlled release of proteins†
Received
19th July 2014
, Accepted 3rd October 2014
First published on 3rd October 2014
Abstract
A general one-pot hydrothermal process was used to prepare different sizes of Fe3O4 nanoparticles dispersed on hydroxyapatite nanorods with CTAB as a surfactant. We also explore the role of hydrothermal reaction temperature and the surfactant on the crystallinity and formation of the rod like morphology of HAp. The obtained nanoparticles are systematically studied by X-ray powder diffraction (XRD), Fourier-transform infrared spectroscopy, Raman spectroscopy, field emission scanning electron microscopy (FESEM) with EDS for elemental mapping, transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET) nitrogen sorptometry and vibrating sample magnetometry (VSM). The as-synthesized Fe3O4/HAp nanocomposites are further explored to study the pH dependent protein adsorption and controlled release using hemoglobin (Hb) as a model protein. A maximum protein adsorption (Qo) of 166.67 mg g−1 is observed for the Fe3O4/HAp nanocomposite and it increases to 200.07 mg g−1 upon increasing the concentration of Fe3O4 nanoparticles. The pH controlled sustained release process is observed for Hb at various pH values of 4.0, 7.4 and 9.0 in phosphate buffer saline (PBS) solution at room temperature. The maximum protein release was obtained for the lower pH values. The dosage dependent in vitro cytotoxicity assays are also performed to confirm biocompatibility of the prepared samples.
1. Introduction
In recent years the synthesis of different morphologies of hydroxyapatite (Ca10(PO4)6(OH)2, HAp) nanostructures have become a topic of extreme interest due to their distinctive properties and potential multifunctional applications. It is an important inorganic bio mineral in bone and teeth.1,2 The HAp nanostructures are fascinating interest in various other applications like nanomedicine, drug and gene delivery, catalyst, gas sensor, biosensor, tissue engineering, adsorption of heavy metals etc.3–6 These applications requires a nanoparticles with uniform size and shape to improve their physicochemical and functional properties. Also, the control of size as well as shape is very much essential to improve the surface area of these nanoparticles for adsorption and drug delivery applications.7–9 Thus the particle geometry plays a key role in improving the functional properties. The HAp crystal structure includes Ca2+, PO43− and OH− ions, the positively charged Ca2+ sites surrounded by negatively charged tetrahedral PO43− units and OH− ions occupy columns parallel to the hexagonal axis. The positively charged Ca2+ ions are mainly present at a (b)-planes and negatively charged PO43− and OH− ions are present in the c-planes.10 These two different charges of HAp may be responsible for the adsorption. The HAp with high specific surface area is prerequisite for an excellent adsorbent properties like higher uptake and release.8,9,11 Also, HAp and its nanocomposites prepared by several techniques have excellent biodegradability, osseointegration, bioactivity, and mechanical properties.5,12 The ZnO and TiO2 doped HAp nanocomposites exhibit an excellent catalytic activity and degrades the industrial dye from waste water under visible light.13 The Ag+ ions incorporated HAp nanocomposite enhances the anti-bacterial activity.14 The lanthanide ions (Eu3+ and Eu3+/Gd3+) incorporated HAp nanorods have potential multifunctional applications like multiple-model imaging agents in magnetic resonance imaging (MRI), computed tomography imaging (CTI) and photoluminescence imaging.15 Strontium (Sr)-doped calcium polyphosphate (SCPP) has been found to improve bone formation and inhibit bone resorption for artificial bone regenerations.16 The noble metals (Au, Ag and Pd, etc.) doped HAp nanocomposites shown biocompatibility and it was used as active catalysts.17 However, very few reports are available for the magnetic nanoparticles incorporated HAp nanocomposites. The magnetic field and magnetic nanoparticles improves the osteoinductivity to accelerate the new bone formation based on these biomaterials.18–24 A number of magnetic nanoparticles are available such as maghemite (γ-Fe2O3), hematite (α-Fe2O3) and magnetite (Fe3O4). Among these the magnetite (Fe3O4) nanoparticles with cubic inverse spinel structure have been extensively used in various biomedical applications such as cell type recognition, magnetic separation, targeted therapeutics and intercellular imaging due to their non-toxic, biocompatibility and unique magnetic properties.25,26 The controlled morphology with narrow size distribution of Fe3O4 nanoparticles were prepared by several colloidal synthetic processes.27–30
The Fe3O4 incorporated HAp or HAp/Fe3O4 nanocomposites are again a promising biomaterials for targeted drug delivery, orthopaedic, hyperthermia-based anticancer treatments, protein adsorption, gene and DNA delivery, novel magnetic guiding for tissue regeneration, heavy metal adsorptions, reusable biosensor and magnetically recyclable/recoverable catalysts etc.19,31,32 In general these nanocomposites can be prepared by various methods such as co-precipitation, dip-coating, ultrasonic spray pyrolysis, biomineralization coating, mechanochemical, electrochemical coating and hydrothermal techniques.20 These processes deliver a wide range of morphologies like nanosheets, nanorods, hollow microspheres, hierarchical mesoporous, plate like nanoparticles and nanotubes.19–24,33–36 Among these, the nanorods exhibit higher specific surface area (SSA) and pore size distributions and it is extensively used as an adsorption of protein and heavy metals from waste water. In general, the cetyltrimethyl ammonium bromide (CTAB) can be used as a soft template for the growth of highly c-axis oriented HAp nanorods.37
Herein, we report two different concentrations of Fe3O4 nanoparticles dispersed on the HAp nanorods prepared by simple hydrothermal process in the presence of CTAB. The influence of reaction temperature and CTAB on the crystallinity and morphology of the samples were also investigated. The nucleation and growth mechanism was systematically examined and proposed a formation mechanism based on the obtained results. Protein adsorption and desorption studies were performed for the prepared nanocomposites with haemoglobin as model protein. The pH and concentration dependent adsorption experiments were also performed for the as-prepared Fe3O4/HAp nanocomposites. The adsorption isotherms for the Fe3O4/HAp nanocomposites were further analysed with the existing mathematical models. The pH controlled protein release studies were conducted at different pH values of 4.5 and 7 in phosphate buffer saline (PBS).24,38–41
2. Experimental section
2.1 Materials
All the reagents and chemicals used in the present investigations were of analytical or equivalent grade and used without further purification. Calcium chloride dihydrate (CaCl2·2H2O), di-ammonium hydrogen phosphate ((NH4)2(HPO4)), ferrous chloride tetrahydrate (FeCl2·4H2O) and ferric trichloridehexahydrate (FeCl3·6H2O) were obtained from Sigma-Aldrich and N-cetyl-N,N,N-trimethyl ammonium bromide (CTAB), ammonium hydroxide (NH4OH), acetone and ethanol were procured from Himedia, India.
2.2 Synthesis of magnetite (Fe3O4) and hydroxyapatite (HAp) nanocomposites by hydrothermal process
The superparamagnetic (Fe3O4) and hydroxyapatite nanocomposite was prepared by hydrothermal process. In a typical synthesis process, 0.01 M of (NH4)2 HPO4 and 0.1 M of CTAB were dissolved in 10 mL of double distilled water with continues stirring. Then, 0.03 M of CaCl2·2H2O was dissolved in 10 mL of water. Further, the CTAB-phosphate solution was added drop wise to the calcium chloride solution and adjusts the pH to10.5 by adding the ammonium hydroxide (30%) with vigorous stirring for 30 min and it is considered as solution A. The same time, 0.01 M of FeCl2·4H2O and 0.015 M of FeCl3·6H2O were dissolved in 10 mL of aqueous solution to maintain the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 for Fe2+ and Fe3+ and the pH was adjusted by ammonium hydroxide (30%). The resultant solution was mixed with solution A and stirred for 20 min to get the translucent mixed solution. This was further transferred to the Teflon-lined stainless steel autoclave and kept in an oven at 180 °C for 12 h. Then it was cooled down to room temperature naturally and the supernatant was discarded by decontanation. The final brown colour precipitate was washed several times with ethanol and water. This was dried at 70 °C under vacuum to obtain the Fe3O4/HAp nanocomposite. This experimental procedure was adopted to prepare the two different molar concentrations (0.5 and 1 M) of iron oxide on HAp. The samples are represented as Fe3O4/HAp-1 for 0.5 mM of Fe3O4 on HAp and Fe3O4/HAp-2 for 1 mM of Fe3O4 on HAp.
:1.5 for Fe2+ and Fe3+ and the pH was adjusted by ammonium hydroxide (30%). The resultant solution was mixed with solution A and stirred for 20 min to get the translucent mixed solution. This was further transferred to the Teflon-lined stainless steel autoclave and kept in an oven at 180 °C for 12 h. Then it was cooled down to room temperature naturally and the supernatant was discarded by decontanation. The final brown colour precipitate was washed several times with ethanol and water. This was dried at 70 °C under vacuum to obtain the Fe3O4/HAp nanocomposite. This experimental procedure was adopted to prepare the two different molar concentrations (0.5 and 1 M) of iron oxide on HAp. The samples are represented as Fe3O4/HAp-1 for 0.5 mM of Fe3O4 on HAp and Fe3O4/HAp-2 for 1 mM of Fe3O4 on HAp.
2.3 Characterization
The X-ray diffraction (XRD) measurements were carried out at room temperature using a PANalytical X'Pert-Pro diffractometer with Cu Kα1 radiation (λ = 1.5406 Å) over a scanning interval (2θ) from 10 to 70°. The average crystallite sizes were estimated using the Scherrer formula from the X-ray line broadening. The infrared spectrum of the samples was obtained by using a Fourier transform infrared (FTIR) spectrometer (Bruker Tensor 27, Germany). The sample was prepared in a KBr pellet for the investigation within the range of 4000 to 450 cm−1. The morphology of the Fe3O4/HAp nanostructures was observed by field emission scanning electron microscopy (FESEM) (FEI Quanta-250 FEG) coupled with EDS spectroscopy and transmission electron microscope (TEM, Hitachi H600) operating at 80 kV. UV-Visible spectral analysis was done by using JoscoV-650 spectrophotometer. The specific surface areas and pore sizes were determined by BET-N2 adsorption for the Fe3O4/HAp nanocrystals using a micromeritics ASAP 2020 surface area analyzer. The magnetic properties were studied using a Lakeshore 7404 vibrating sample magnetometer (VSM) at room temperature.
2.4 In vitro protein adsorption
Batch mode adsorption experiments were carried out to determine the maximum loading capacity of the protein on nanocomposites. Hemoglobin (Hb) was used as a model protein for the present investigation. The Hb adsorption experiment with various concentrations was executed by the following process. Initial experiments were conducted to evaluate the effect of pH (pH 4–10) for loading the Hb on two different adsorbents (Fe3O4/HAp-1 and Fe3O4/HAp-2). The experimental processes were conducted separately for the two adsorbents. The powdered nanoparticles of 5 mg were immersed in phosphate buffer saline (PBS) solution with the protein concentration of 2 mL, 500 μg mL−1. The pH of the samples was adjusted between 4 to 10 using 0.1 M HCl and NaOH solutions. All the solutions were shaken at a constant room temperature for 4 hours. The final solution was centrifuged and the supernatant was taken out and measured the UV-vis absorption spectrum at a wavelength of 405 nm. The maximum Hb adsorption was observed at pH 7 for the Fe3O4/HAp-1 and Fe3O4/HAp-2 samples and it was chosen as the optimum pH for entire successive adsorption experiments. The adsorption studies were performed by varying the protein concentration ranging from 500 to 2500 μg mL−1 at pH 7. The protein adsorption capacity of the nanocomposites was calculated by the following mathematical equations.| |
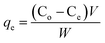 | (1) |
where qe is the Hb adsorption capacity on unit amount of the nanocomposites (mg g−1). The Co and Ce is the initial Hb concentration and final or equilibrium Hb concentration of protein solution (μg mL−1). V is the volume of Hb solution (mL) and W is the dry weight of the nanocomposites (mg).
2.5 In vitro pH controlled protein release
The pH dependent controlled release of Hb was carried out by immersing a50 mg of Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites in 20 mL of Hb in phosphate buffer saline (PBS) solution with the concentration of 2000 μg mL−1 at pH 7. This solution was shaken at room temperature for 4 hand followed by centrifugation and freeze drying to obtain the Hb-Fe3O4/HAp-1 and Hb-Fe3O4/HAp-2 for drug delivery. The in vitro protein release experiments with various pH values were executed as follows. The Hb loaded nanoparticles of 8 mg was immersed in 8 mL of PBS solution and adjusted the pH values as 4, 7.4 and 9 at room temperature with constant shaking of 120 rpm for 4 h. The 2 mL of Hb release solution was withdrawn for pre-determined intervals and replaced with the same volume and pH of fresh PBS solution. The absorption rate of Hb release was measured by UV-vis absorption spectroscopy at a wavelength of 405 nm.
2.6 In vitro cytotoxicity studies
The cytotoxicity of the Fe3O4/HAp nanocomposites was evaluated by MTT (3-(4,5-dimethylthiahiazol-2-yl)-2,5-diphenyltetrazolium) assay. The human gastric carcinoma cells (MGC-803) was used for the cell viability studies. The MGC-803 was cultured in a RPMI-1640 medium supplemented with 20% fetal bovine serum and 2% penicillin–streptomycin for 24 hours at 37 °C. The MGC-803 cells were first seeded into 96-well plates with a density of 1 × 104 cells per well and incubated for 48 h. The cells were then treated with different dosage of Fe3O4/HAp nanoparticles (50–300 μg mL−1) for 24 h at 37 °C and the untreated cells served as the control. After incubation, the Fe3O4/HAp nanocomposites were added into wells at treated with various dosages from 50 to 300 μg mL−1 and co-cultured with cells for 24 hours. The cell viability percentage was calculated by the ratio of absorbance of triplicate readings with control wells and the experiments were repeated three times and the mean viability of the three ±standard deviation was determined.
3. Results and discussion
The X-ray diffraction (XRD) patterns were used to analyse the crystalline nature and identify the phases present in the as-prepared nanocomposites. The XRD patterns for the nanocomposite indicates the dual phases of pure HAp (JCPDS card: no. 09-0432) and Fe3O4 (JCPDS card: no. 89-0688), as shown in Fig. 1a and b. The major diffraction peaks appears at 25.87, 31.42, 32.11, 32.52, 39.62 and 49.42° corresponds to the (002), (211), (112), (300), (202), (310), (222), (213) and (004) crystal planes of HAp and 35.59 and 43.83° for the (311) and (400) crystal planes of Fe3O4 respectively. The increase in the concentration of iron oxide notably enhances the relative intensity of (311) plane and there is no significant shift in the peak positions of HAp as shown in Fig. 1b. It indicates the incorporation of Fe3O4 nanoparticles does not influence the crystalline nature and phase purity of HAp. This clearly confirms the formation of well crystallized hexagonal phase of HAp and cubic spinal phase of Fe3O4 without any impurity phases.
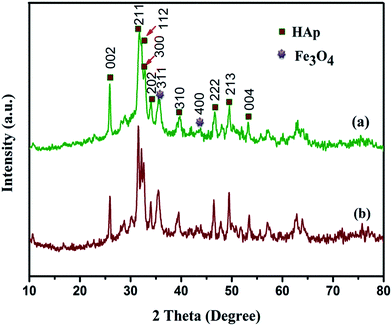 |
| | Fig. 1 XRD patterns for (a) Fe3O4/HAp-1 (0.5 mM of Fe3O4) and (b) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
The FTIR spectra for Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites are shown in Fig. 2. The adsorption bands observed at 471, 564 and 604 cm−1 corresponds to the bending vibration (ν4) of phosphate groups. The (ν1) vibration of phosphate was observed at 962 cm−1. The absorption bands at 1044 and 1092 cm−1 is attributed to the stretching vibration (ν3) of phosphate group. The sharp absorption peaks at 3570 and 634 cm−1 corresponds to the stretching of an OH band which confirms the incorporation of iron oxide could not affect the OH site of HAp. The broad absorption bands at 3447 and 1639 cm−1 is due to the H2O present in the synthesized samples.
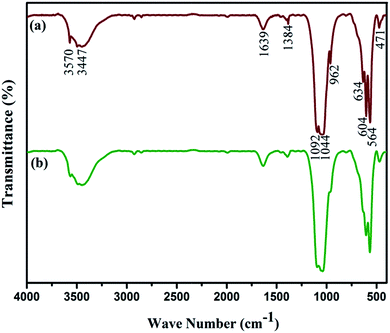 |
| | Fig. 2 FTIR spectra for (a) Fe3O4/HAp-1 (0.5 mM of Fe3O4) and (b) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
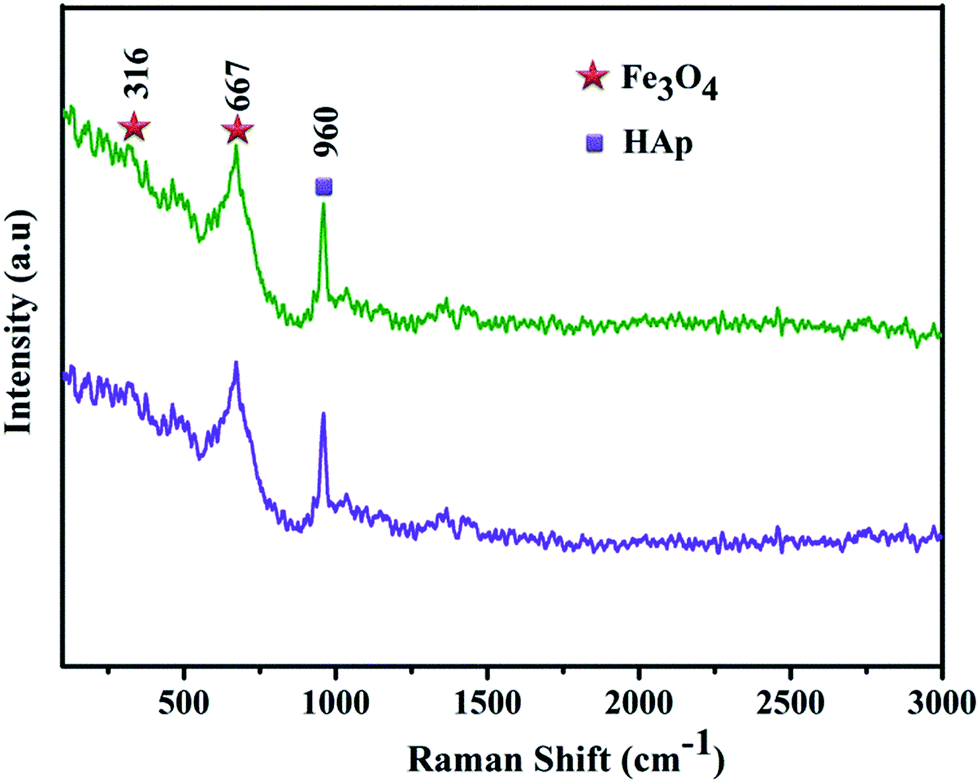
Further, it confirms that there is no significant peak shift was observed by increasing the concentration of iron oxide and also the incorporation of Fe3O4 does not influences the structure of HAp. The Raman spectroscopy has been used to complete the structural analysis of Fe3O4/HAp nanocomposite for the two different concentrations of iron oxide. The magnetite (Fe3O4) has a cubic inverse spinel structure with the iron ions (Fe3+ and Fe2+) occupy interstitial tetrahedral and octahedral sites and the oxygen ions form an fcc closed packed structure symbolized as [Fe3+]–[Fe2+Fe3+]2O4. The magnetite crystal with the space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m has five Raman active modes such as A1g, three T2g, and Eg, and four IR active bands (T1u).
m has five Raman active modes such as A1g, three T2g, and Eg, and four IR active bands (T1u).
In the centro-symmetrical space group Fdm involves mutual exclusion of IR and Raman vibrational modes. The Fe3+ and O2− ions occupy the Td and C3v sites respectively and it contribute to the Raman activity, although bivalent iron cations are not directly involved.
A broad intense peak at 669 cm−1 in Fig. 3a and b can be ascribed to the active mode of magnetite A1g and less intense peaks at 540 and 309 cm−1 corresponds to the Raman active modes of T2g, and Eg. In addition, the characteristic band at 960 cm−1 assigned to the symmetric stretching (ν1) of the phosphate groups (PO43−). These Raman spectra again confirm the Fe3O4 does not influences the crystal structure of Fe3O4/HAp nanocomposites. The morphology and elemental compositions of the as-prepared nanocomposites for the two different molar concentrations of Fe3O4 (0.5 and 1.0 mM) are investigated by FESEM with EDX mapping and TEM. Initially, the Fe3O4 solution is prepared by co-precipitation technique with Fe2+ and Fe3+ ions in the molar ratio of 1:2 with the total molar ratio of 0.5 mM. Further, the total molar ratio of iron ions (Fe2+ + Fe3+) was increases to 1 mM. The size of the synthesized Fe3O4 nanocrystals increases from 12 to 27 nm by increasing the concentration from 0.5 to 1 mM as shown in (ESI) Fig. 1a and b.† The concentration of iron ions strongly influences the nucleation growth rate of Fe3O4 nanocrystals. The higher concentration of iron ions led to increase the formation of large numbers of seed nuclei and attaching together to form a bigger size Fe3O4 nanoparticles. The typical FESEM images of the as-prepared nanocomposites with two different Fe3O4 concentrations dispersed on HAp are shown in Fig. 4a–d. It shows the 0.5 mM of Fe3O4 nanoparticles dispersed on HAp exhibits rod like morphology with slightly ellipsoidal shape. The size of the nanorods in Fig. 4a and b shows an average diameter and length of 82 and 423 nm respectively for the 12 nm size of Fe3O4 nanoparticles homogeneously dispersed on the HAp nanorods.
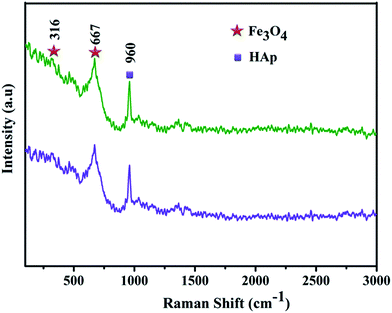 |
| | Fig. 3 Raman spectra for (a) Fe3O4/HAp-1 (0.5 mM of Fe3O4) and (b) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
 |
| | Fig. 4 FESEM images for (a and b) Fe3O4/HAp-1 (0.5 mM Fe3O4) and (c and d) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
Fig. 4c and d shows the morphology of the 1 mM of Fe3O4 dispersed on HAp nanocomposite. It shows the rod like morphology with the diameter and length of 39 and 350 nm respectively. This confirms the large number and higher diameter of Fe3O4 nuclei controls the nucleation growth which significantly reduces the diameter and length of the HAp nanorods. Also, the narrow sized Fe3O4 nanoparticles are uniformly distributed on the surface of the HAp nanorods. Further, the dispersion of magnetite nanoparticle on the HAp nanorods was clearly visible from the TEM micrographs. Fig. 5a and b shows TEM image for 0.5 mM Fe3O4 magnetic nanoparticles distributed on the surfaces of the HAp nanorods with smaller size. Moreover, the image in Fig. 5c and d shows the increased size and density of the Fe3O4 nanoparticles by increasing the Fe3O4 concentration to 1 mM and it also distributed on the surface of HAp nanorods. Further, FESEM-EDS and elemental mapping in ESI Fig. 2a–f† provides the clearer information about distribution of elements in the nanocomposites. It indicates the samples are composed of calcium (Ca), phosphorus (P), oxygen (O) for the HAp template phase and the iron (Fe) for Fe3O4 nanoparticles on the surface of HAp nanorods. The increase in iron concentrations from 0.5 to 1.0 mM in the nanocomposites shows the higher numbers of Fe species were dispersed on the surface of HAp as seen in ESI Fig. 2f.†
 |
| | Fig. 5 TEM micrographs for (a and b) Fe3O4/HAp-1 (0.5 mM Fe3O4) and (c and d) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
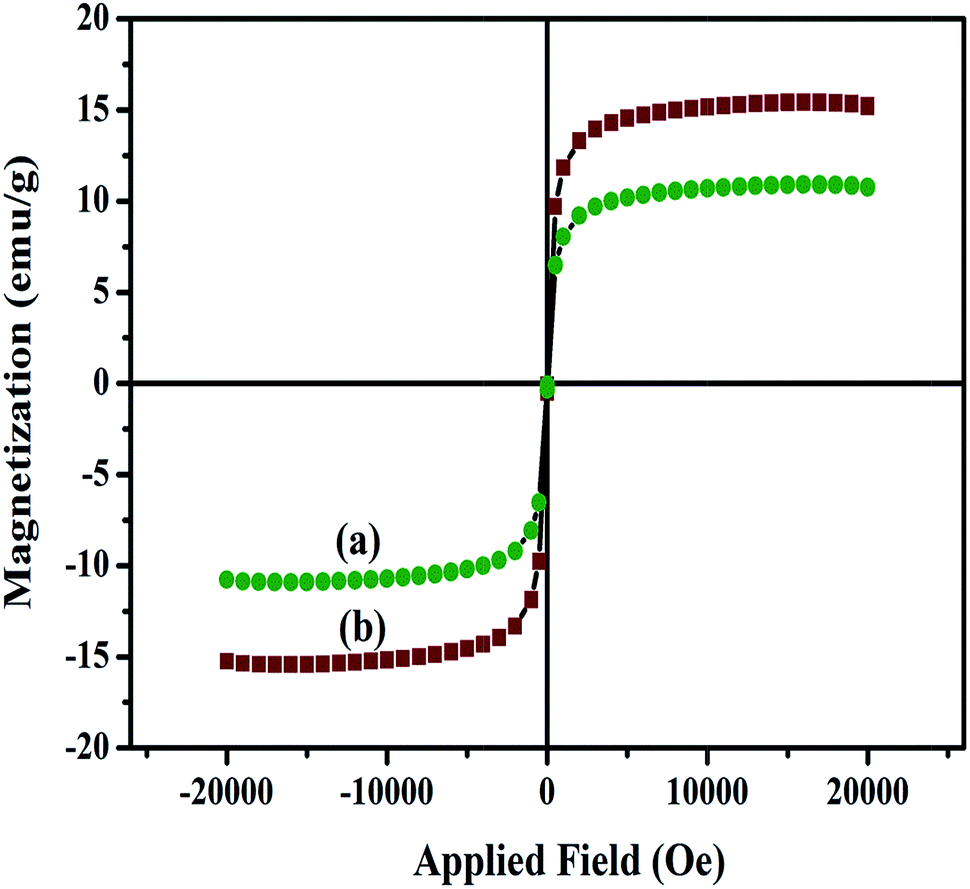
The magnetic properties of the nanocomposites have also been investigated at room temperature using a vibrating sample magnetometer (VSM) with an applied magnetic field of −2 to +2 kOe. The hysteresis loops for the Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites in Fig. 6 shows the superparamagnetic behaviour at room temperature. The saturation magnetization (Ms) obtained from the hysteresis loop for the HAp/Fe3O4-1 and HAp/Fe3O4-2 nanocomposites are 11.5 and 15.5 emu g−1 respectively with zero coercivity (Hc). Generally, the increase in the particles size increases the saturation magnetization and coercivity with in the superparamagnetic region. The increase in the average grain size from 12 to 27 nm for the two different concentrations of Fe3O4 nanoparticles grown on the HAp nanorods may be possible increase of the magnetization. The surface area and pore size distribution of HAp/Fe3O4 nanocomposites are studied by nitrogen physisorption (adsorption–desorption) experiments. Fig. 7a and b shows the BET surface area and their corresponding BJH plot for pore size distribution. The adsorption–desorption isotherms confirm a type IV isotherm loop at a relative pressure between 0 to 1. Both the samples show mesopores with the pore size distribution of 5–20 and 15.3 nm for the HAp/Fe3O4-1 and HAp/Fe3O4-2 nanocomposites using Barrett–Joyner–Halenda (BJH) calculations.
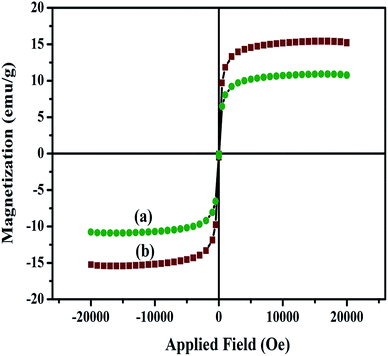 |
| | Fig. 6 Magnetic hysteresis loops for (a) Fe3O4/HAp-1 (0.5 mM Fe3O4), (b) Fe3O4/HAp-2 (1.0 mM Fe3O4) nanocomposites measured at room temperature. | |
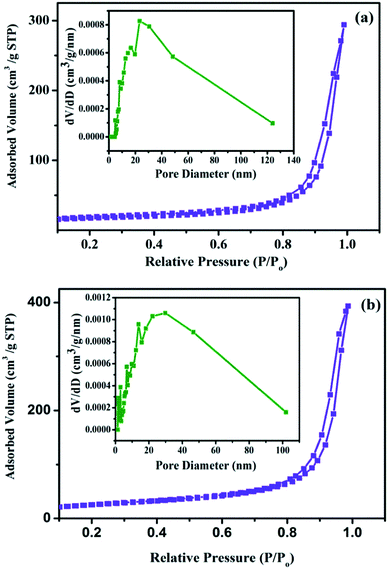 |
| | Fig. 7 Nitrogen adsorption–desorption isotherms and pore-size distribution (inset) for (a) Fe3O4/HAp-1 (0.5 mM Fe3O4) and (b) Fe3O4/HAp-2 (1 mM of Fe3O4) nanocomposites. | |
The obtained specific surface areas for the HAp/Fe3O4-2 and HAp/Fe3O4-1 are 91.2 about 80.7 m2 g−1 respectively. The higher surface area of HAp/Fe3O4-2 can be due to the increase of iron oxide concentration decreases the grain size of the HAp. This smaller grain size of the HAp enhances the specific surface area. The grain size obtained from XRD and the FESEM images also supports the HAp/Fe3O4-2 has small grains as well as the particle sizes required to increase the specific surface area with porous structure.
3.1 Nucleation growth mechanism
The formation mechanism of two different concentrations of Fe3O4 nanoparticles dispersed on the highly c-axis oriented one dimensional HAp nanorods are shown in Fig. 8 based on the obtained results. The Fe3O4/HAp with two different concentrations of iron oxide (0.5 and 1.0 mM) were prepared by hydrothermal process with CTAB as surfactant. The hydrothermal temperature and CTAB plays a key role in improving the crystallinity and growth. During the growth process, cationic CTAB was dissolved in deionized water and forms spherical shaped micelle. The increase in CTAB concentrations above the critical micelle concentration (CMC) of 1 mM, resulting the formation of rod like micelles by the combination of spherical shape CTAB micelles. Subsequently the tetrahedral phosphate (PO43−) ions are attached with the rod shaped tetrahedral CTAB micelle via electrostatic interaction and form a pre-nucleation growth site for the formation of 1D HAp. In addition, the Ca2+ ions are also slowly incorporated to the above solution. The Ca2+ ions are deposited and nucleation growth starts along the c-axis and forms the rod shaped hydroxyapatite nanostructures. In addition, the hydrothermal reaction temperature also plays a vital role in the crystallinity and shape evaluation of HAp nanorods. Further, the magnetically induced HAp nanorods are prepared by the addition of certain molar ratios (Fe2+ + Fe3+ = 0.5 and 1 mM) of Fe2+ and Fe3+ ions (1:2) during the crystallization process of HAp. The increase in the total molar ratio of Fe2+ and Fe3+ ions from 0.5 to 1 mM increases the size of the synthesized Fe3O4 nanocrystals from 12 to 27 nm. This maybe due to the increase in the concentration of iron ions strongly develop the nucleation and growth rate of Fe3O4 nanocrystals. The increase in the iron ions concentration from 0.5 to 1 mM increases the formation of a large number of seed nuclei. It provides a larger diameter of Fe3O4 nanoparticles which are dispersed on the 1D HAp nanostructures. The concentration dependent size of the Fe3O4 nanoparticles dispersed on the HAp nanorods can be seen through the FESEM and TEM micrographs and EDX mapping. The concentration dependence of the iron ions are further confirmed from the change in the intensity of the (311) peak from the XRD pattern and it increases with the increasing concentration. The magnetic properties also changes for the two different concentrations of Fe3O4 nanoparticles. The specific saturation magnetization (Ms) obtained from hysteresis loop increases from 10.6 to 15.5 emu gm−1 for the 0.5 and 1 mM Fe3O4 concentrations.
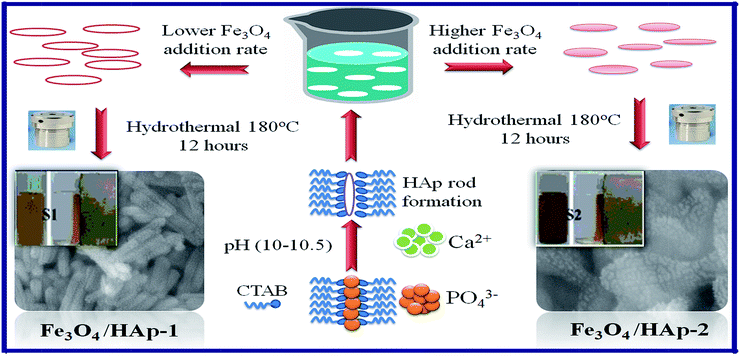 |
| | Fig. 8 Schematic illustration for the formation mechanism of two different concentrations of Fe3O4 nanoparticles dispersed on 1D HAp nanorods. | |
The larger size of Fe3O4 nanoparticle dispersed on HAp (HAp/Fe3O4-2) nanocomposite shows higher Ms due to the surface effect and increased concentration of Fe3O4 nanoparticles. Meanwhile, the larger size Fe3O4 nanoparticle reduces the nucleation growth of HAp which reduces the size of the HAp nanorods. The size reduction in HAp increases the specific surface area. Thereby, these nanobiomaterials will be efficiently used for various biomedical applications like drug delivery, gene delivery, protein adsorption and potential adsorbents.
3.2 Adsorption behavior of hemoglobin (Hb) on two different concentrations of Fe3O4 dispersed on HAp nanoparticles
The adsorption behavior of hemoglobin on two different concentrations of Fe3O4 nanoparticles dispersed on HAp nanorods have been systematically evaluated based on the effect of pH, initial concentration of Hb and function of time. The initial pH of the solution is an essential parameter influences the rate of loading of Hb in the nanocomposite.
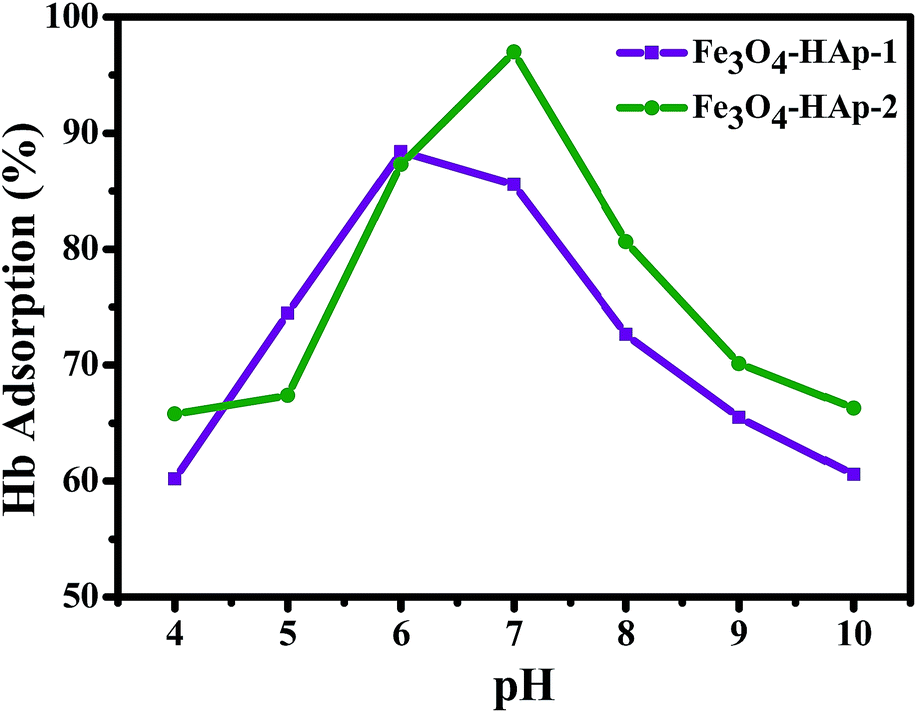
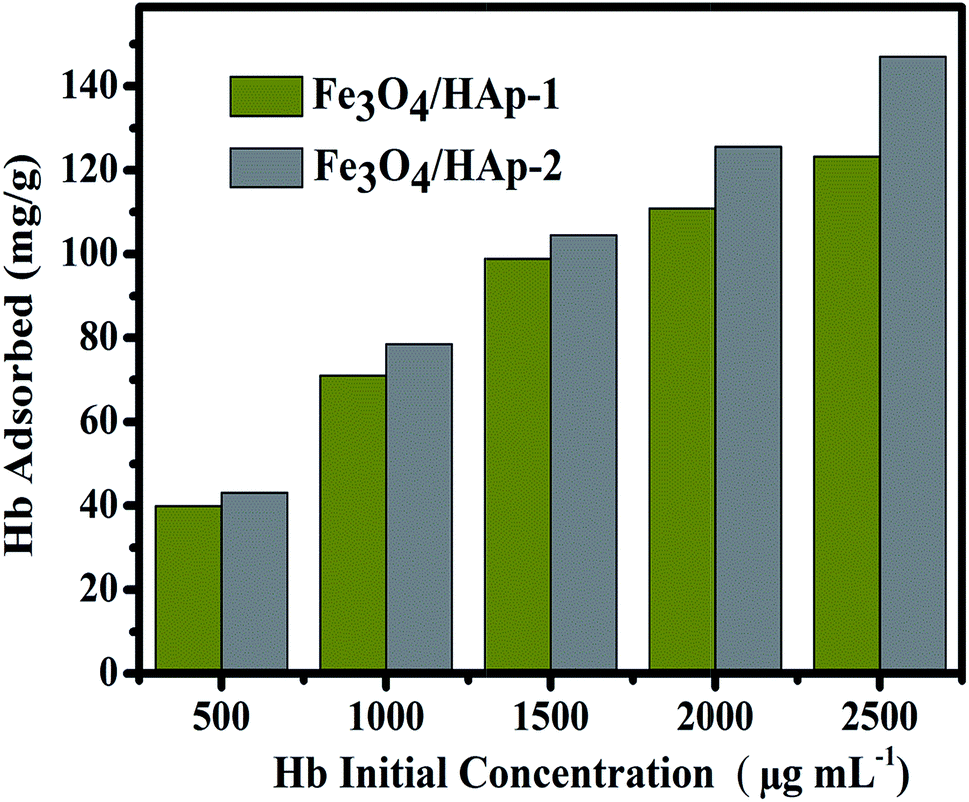
The Hb is a neutral charge protein and Fe3O4/HAp is more positive charged nanoparticles than the pure HAp. The increase in Fe3O4 concentrations significantly increases the positive entities and enhances the loading capacity of protein. Adsorption of Hb on Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites was conducted with the range of pH from 4 to 10 with the initial Hb concentration of 500 μg mL−1. Fig. 9 shows the pH dependent Hb adsorption ability of the nanocomposites with an initial Hb concentration of 500 μg mL−1 in PBS. The maximum Hb adsorption obtained for the Fe3O4/HAp-1 nanocomposite is 85% at pH 6.
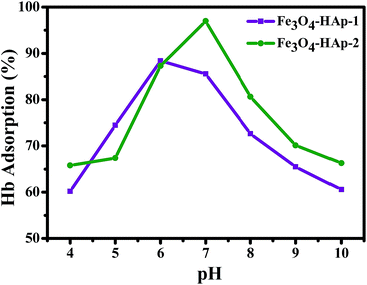 |
| | Fig. 9 The pH dependent adsorption (%) of hemoglobin (Hb) on the Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites with an initial Hb concentration of 500 μg mL−1 in PBS solution. | |
The other Fe3O4/HAp-2 nanocomposite exhibits the maximum adsorption of 95% at pH 7 near the isoelectric point of the protein (pI(Hb) = 6.8). De Bruin et al. (1969) have already reported that the different pH of 5.75, 6.7 and 7.4 of Hb have different charges of +13, +5 and −1 mV. In the present case, maximum adsorption percentage obtained for the nanocomposites in the pH range of 7–8. Since the negative charge of the protein leads to a strong electrostatic interaction with Fe3O4/HAp nanocomposites. Meanwhile, the maximum adsorption percentage was obtained at pH 7 for the increased concentration of Fe3O4. The electrostatic repulsion occurs between different the Fe3O4 nanoparticles in Fe3O4/HAp nanocomposite and Hb by increasing the pH to 10 due to the identical net charge and reduces the loading percentage of Hb. The higher percentage of Hb adsorption occurs for the nanocomposites occurs between the pH of 6–7. Thereby, the further adsorption experiments are conducted with the pH of 7. The Hb loading capacity of the as-prepared nanocomposite was investigated by varying the initial concentrations of Hb from 500 to 2500 μg mL−1 and the experimental results are shown in Fig. 10. It shows the loading capacity of the as-prepared nanocomposites increases with increasing initial concentration of Hb. The loading capacity of the Fe3O4/HAp-2 nanocomposite reaches a maximum of 150 mg g−1 for the adsorbed Hb at an initial concentration of 2500 μg mL−1. This adsorption capacity was slightly higher than that of 120 mg g−1 for the Fe3O4/HAp-1 nanocomposite. These results show the as-prepared nanocomposites can exhibit an excellent protein adsorption for the application of nano drug carrier.
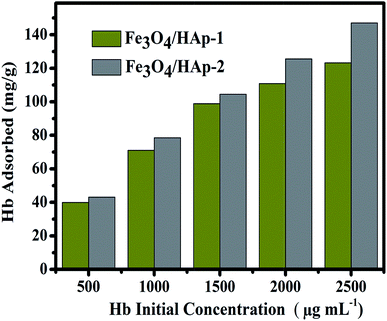 |
| | Fig. 10 The Hb loading capacity of the as-prepared Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites with different initial protein concentration in the range of 500 to 2500 μg mL−1. | |
3.3 Adsorption isotherms
The adsorption isotherms were further analyzed based on the existing mathematical models. These models describe the adsorption capacity of Hb on the nanocomposites and study their nature of adsorption. The characteristic adsorption constants were estimated by fitting the experimental data using Langmuir and Freundlich models to understand the adsorption capacities of Hb on the nanocomposites. The adsorption isotherms of Hb on the two nanocomposites are shown in Fig. 11a and b and these data are fitted based on Langmuir and Freundlich isotherms to explain the adsorption performances. The appearance for the Langmuir and Freundlich isotherm is based on the following two equations42| |
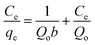 | (2) |
| |
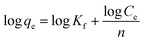 | (3) |
where qe and Ce are the equilibrium concentrations of protein (μg mL−1) on adsorbent and in solution respectively. Qo and b is Langmuir constants related to adsorption capacity and energy of adsorption respectively. The value of Qo and b indicates the maximum adsorption capacity corresponds to a saturated monolayer of proteins on adsorbent surface. The Freundlich isotherm model undertakes heterogeneous surface energies in which the energy term in Langmuir equation varies as a function of surface coverage and Kf and 1/n are Freundlich constants associated to adsorption capacity and intensity of the adsorption respectively.
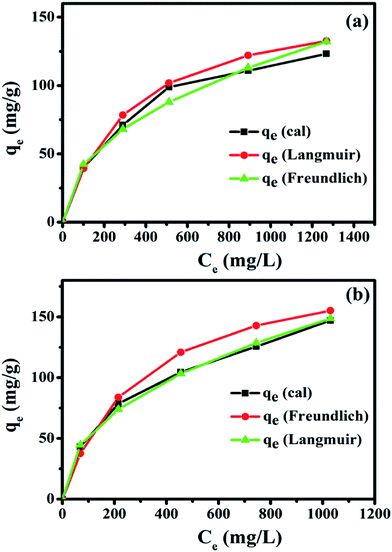 |
| | Fig. 11 Adsorption isotherms of Hb on the nanocomposites with Langmuir and Freundlich models for (a) Fe3O4/HAp-1 and (b) Fe3O4/HAp-2 in PBS buffer at pH 7.4. | |
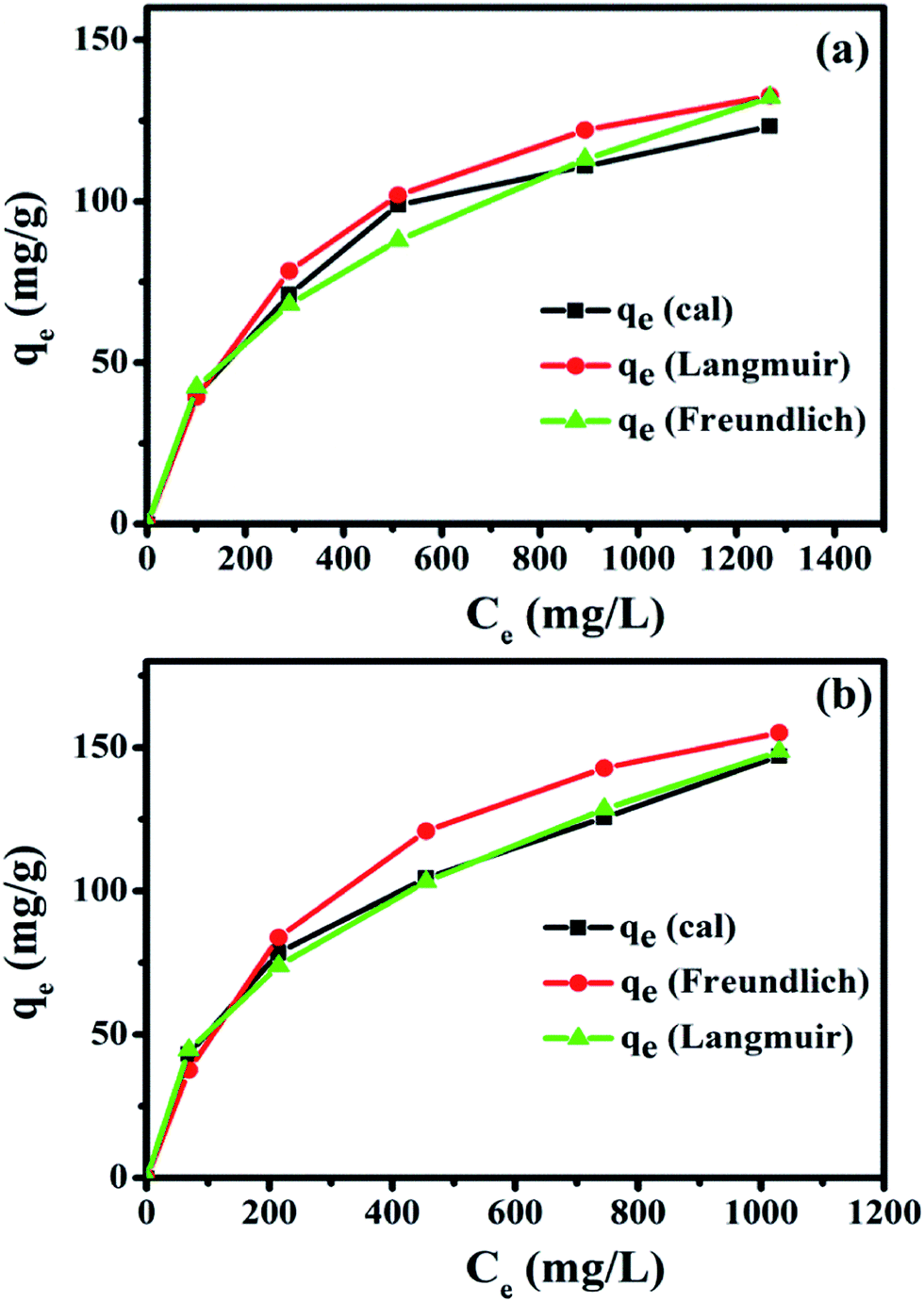
The relationship between the adsorption capacity and initial Hb concentration was observed by both Langmuir and Freundlich isotherms. The adsorption isotherms of Hb on Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites were determined in the concentration range of 500–2500 μg mL−1. The observed experimental results in Fig. 11 shows the increase in the initial concentration of Hb increases adsorption rate on the Fe3O4/HAp-1. It is observed that the Hb adsorption is higher for the Fe3O4/HAp-2 at the same initial protein concentrations. Both the experimental data are fitted with Langmuir and Freundlich isotherms for the maximum adsorption capacity of Hb on the nanocomposites and the fitted parameters are summarized in Table 1. The correlation coefficient (R2) of Fe3O4/HAp-1 nanocomposite exhibits 0.9972 for Langmuir and 0.9723 for the Freundlich isotherms and the maximum adsorption capacity (Qo) of the sample is 166.67 mg g−1. The correlation coefficient (R2) of Fe3O4/HAp-2 nanocomposite is 0.9923 for Langmuir and 0.9841 for the Freundlich isotherms and the maximum loading capacity (Qo) of the sample is 200 mg g−1. Further, the Langmuir model was used to calculate the Hb adsorption and it indicates the monolayer adsorption with the maximum adsorption occurs for the Fe3O4/HAp-2 than that of Fe3O4/HAp-1 nanocomposites.
Table 1 Fitted parameters for the Langmuir and Freundlich models of adsorption isotherm for the Hb loaded on Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites
| Samples |
Langmuir parameters |
Freundlich parameters |
| R2 |
Qo (mg g−1) |
b (1 mg g−1) |
R2 |
Kf (mg g−1) |
1/n |
| Fe3O4/HAp-1 |
0.9972 |
166.67 |
0.0031 |
0.9723 |
5.3456 |
0.4490 |
| Fe3O4/HAp-2 |
0.9841 |
200.01 |
0.0033 |
0.9946 |
6.7765 |
0.4450 |
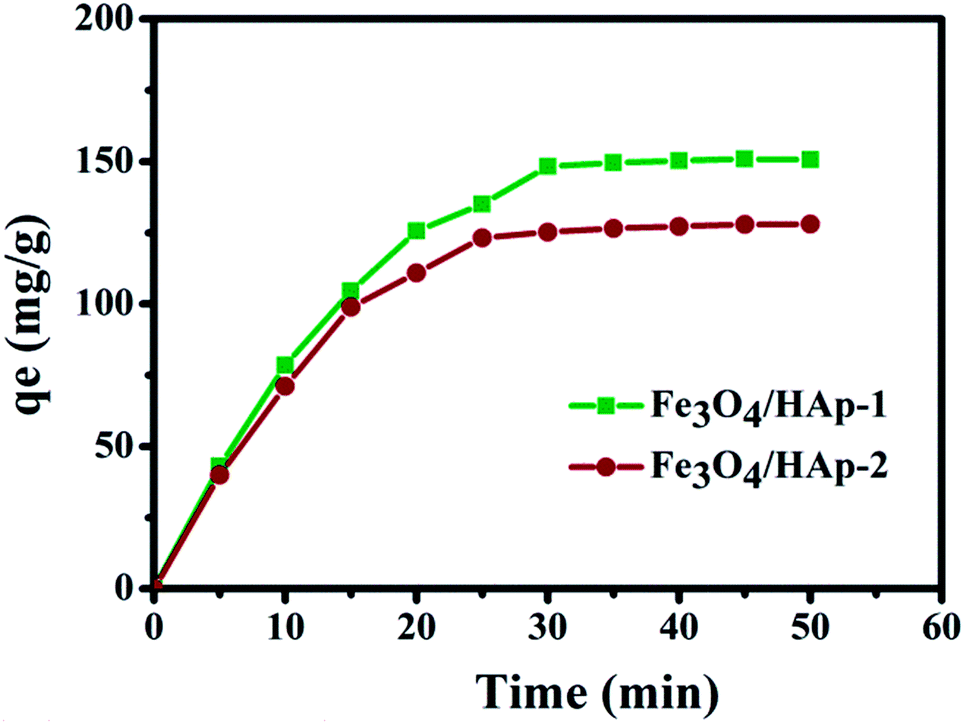
The regression values of both nanocomposites clearly confirm the experimental data was best fitted for the Langmuir isotherm than that of the Freundlich isotherm. It concludes that the adsorptions of Hb on the nanocomposites may be monolayer adsorption on a surface with homogeneous system. Subsequently, the Hb adsorption capacity of the HAp nanorods was higher than that of the available reported values as presented in Table 2. The adsorption kinetics of Hb on the nanocomposites in Fig. 12 shows the protein adsorption process is a time dependent phenomena. Also, the present nanocomposite materials show excellent and fast adsorption rate. The Fe3O4/HAp-1 nanocomposite exhibit an adsorption capacity of Hb and the rate of adsorption rapidly increases for the initial 20 min and reach the equilibrium after 25 min. But, in the case of Fe3O4/HAp-2 reaches the equilibrium only after 30 min. The maximum adsorption of Hb on the nanocomposites increases with increase in adsorption time. Therefore, the contact and equilibrium times play an important role in the adsorption of proteins and the loading capacity can be controlled by varying the time.
Table 2 Summary of the maximum Hb adsorption capacities of different biomaterials
| S. no |
Biomaterials |
Maximum adsorption capacity Qo (mg g−1) |
Reference |
| 1 |
Polyhedral like hydroxyapatite |
164 |
11 |
| 2 |
Hydroxyapatite (HAp)/Fe3O4 |
150 |
24 |
| 3 |
HAP porous hollow microspheres |
120 |
39 |
| 4 |
Hierarchical HAp |
177 |
41 |
| 5 |
Mesoporous silica |
57 |
43 |
| 6 |
Fe3O4@NH2 |
34.51 |
44 |
| 7 |
Fe3O4/HAp-1 |
166.67 |
Present study |
| 8 |
Fe3O4/HAp-2 |
200.01 |
Present study |
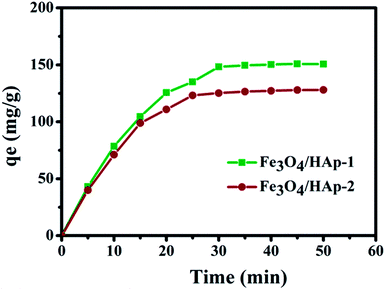 |
| | Fig. 12 Adsorption kinetics of Hb on the Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites and the initial concentration of Hb 500 μg mL−1 in PBS buffer at pH 7.4. | |
3.4 pH controlled release of Hb
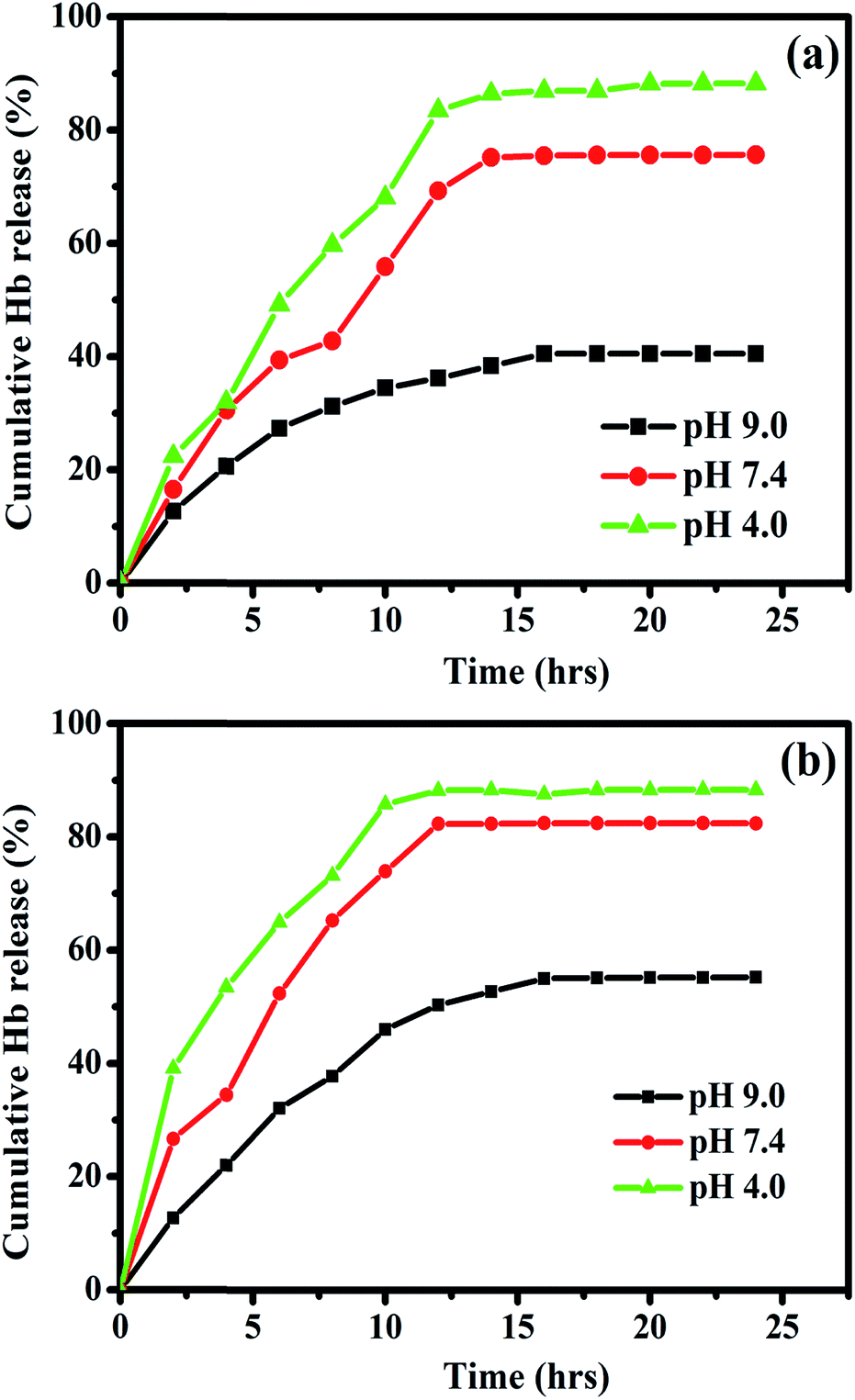
The releasing process was evaluated for the Hb loaded nanocomposites with different pH values in PBS at room temperature. The pH dependent cumulative release ratio (%) is given by the amount of Hb released in PBS solution as a function of time. Three different pH values of 9, 7.4 and 4 are chosen for the pH dependent release of Hb. Fig. 13a and b shows the controlled release profile for the Hb loaded Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites. The release profile confirms the Hb adsorption of both nanocomposites exhibits a slow and sustained release over a period of 25 h. The release percentage of Hb increases for the first 13 h for all the pH and attain a steady release till 25 h. The amount of Hb release in PBS with the pH of 4.0 is much larger than that of other two pH values of 7.4 and 9. Therefore the releasing of Hb from Fe3O4/HAp is higher at the acidic condition with the pH of 4 due to the maximum release of calcium and phosphate ions in the range of 41 and 62 mg L−1.37 Also, the cumulative Hb release percentages (%) in PBS solution with a pH value of 4 reaches a maximum of 87 and 95% corresponds to Fe3O4/HAp-1 and Fe3O4/HAp-2 respectively. Thus, the dissolution of hydroxyapatite enhances the protein release in PBS at lower pH values. These experiments confirm the Fe3O4/HAp nanocomposites are excellent and promising biomaterials as potential pH sensitive drug carrier. The present nanocomposites are an excellent pH sensitive drug carrier and it is potential nano biomaterial for tumour and cancer cell treatments.
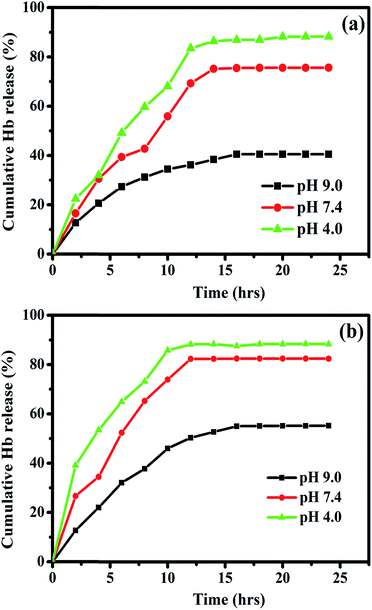 |
| | Fig. 13 Cumulative release of hemoglobin (Hb) loaded on (a) Fe3O4/HAp-1 and (b) Fe3O4/HAp-2 nanocomposites at different initial pH values of 4, 7.4 and 9 in PBS at room temperature. | |
3.5 In vitro cytotoxicity studies
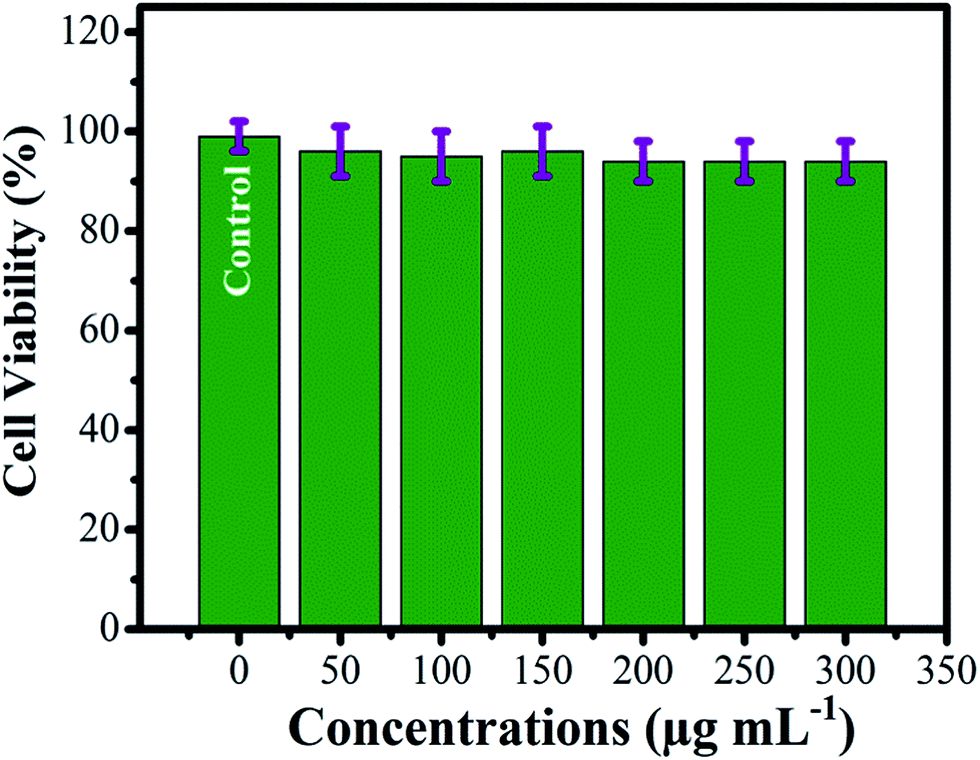
The dose dependent cytotoxicity analysis for the Fe3O4/HAp nanocomposite was performed by using human gastric carcinoma cells (MGC-803). The MTT assay was performed as a colorimetric assay to measure cytotoxicity of the nanocomposites. The observed results for the Fe3O4/HAp nanocomposite exhibits 99.5% of cell viability at the concentration of 50 μg mL−1 as shown in Fig. 14. Interestingly, the higher dosage of 200 μg mL−1 shows the cell viability of 95.4%. Also, the increasing concentration of Fe3O4/HAp nanocomposite between 200 and 300 μg mL−1 shows the similar cell viability of 95.4%. These results confirm the Fe3O4/HAp nanocomposites have modest cytotoxic effect on human gastric carcinoma cells (MGC-803). Hence, we conclude these nanocomposites are biocompatible and promising biomaterials for biomedical applications such as tumor treatment, drug delivery, protein adsorption etc.
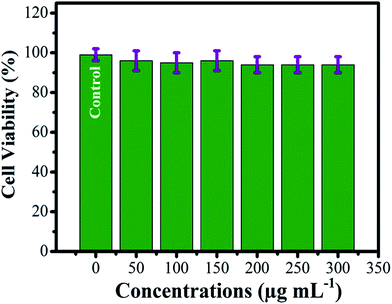 |
| | Fig. 14 Cell viability of Fe3O4/HAp nanocomposite with human gastric carcinoma cells (MGC-803) and cell cultured with different dosages of the samples between 50 and 300 μg mL−1. | |
4. Conclusion
We successfully prepared the nanocomposites of Fe3O4 on HAp nanorods by in situ hydrothermal process for two different concentrations of Fe3O4. The soft template CTAB plays an important role in the formation of nanorods and the reaction temperature improves the crystallinity of Fe3O4/HAp nanocomposites. The morphological analysis confirms the dimensions of the HAp nanorod decreases by increasing the concentration of Fe3O4. The higher grain size of Fe3O4 controls the nucleation growth of HAp. The pH dependent Hb adsorption and sustained release of these nanocomposites clearly indicates their excellent controlled release and the maximum release was obtained at pH 4. The experimental data was best fitted with Langmuir models with the maximum adsorption capacity of 166.67 and 200.07 mg g−1 corresponding to the Fe3O4/HAp-1 and Fe3O4/HAp-2 nanocomposites. Thus, these nanocomposites are excellent biomaterials to be useful in biomedical applications such as tumor treatment, drug delivery, protein adsorption etc.
Acknowledgements
The authors would like to acknowledge the University Grant Commission (UGC), New Delhi, Government of India for the financial support through the major research project and DST-PURSE, Government of India for the FESEM facility.
Notes and references
- H. Monma and T. Kamiya, J. Mater. Sci., 1987, 22, 4247–4250 CrossRef CAS.
- K. S. Katti, D. R. Katti and R. Dash, Biomed. Mater., 2008, 3, 034122 CrossRef PubMed.
- D. A. Wahl and J. T. Czernuszka, Eur. Cells Mater., 2006, 11, 43 CAS.
- S. V. Dorozhkin, J. Mater. Sci., 2007, 42, 1061 CrossRef CAS.
- G. Xiaohui, W. Wang, W. Guolong, J. Zhang, C. Mao, Y. Deng and H. Xia, New J. Chem., 2011, 35, 663–671 RSC.
- G. Bharath, A. Jagadeesh Kumar, K. Karthick, D. Mangalaraj, C. Viswanathan and N. Ponpandian, RSC Adv., 2014, 4, 37446–37457 RSC.
- K. Kandori, S. Toshima, M. Wakamura, M. Fukusumi and Y. Morisada, J. Phys. Chem. B, 2010, 114, 2399–2404 CrossRef CAS PubMed.
- W. H. Lee, C. Y. Loo, K. L. Van, A. V. Zavgorodniy and R. Rohanizadeh, J. R. Soc., Interface, 2012, 9, 918–927 CrossRef CAS PubMed.
- Q. Chao, Z. Ying-Jie, L. Bing-Qiang, X. Y. Zhao, J. Zhao, F. Chen and J. Wu, Chem.–Eur. J., 2013, 19, 5332–5341 CrossRef PubMed.
- C. Chen, Z. Huang, W. Yuan, J. Li, X. Cheng and R. Chi, CrystEngComm, 2011, 13, 1632–1637 RSC.
- X. Y. Zhao, Y. J. Zhu, F. Chen, B. Q. Lu and J. Wu, CrystEngComm, 2013, 15, 206–212 RSC.
- J. S. Cho, D. S. Yoo, Y. C. Chung and S. H. Rhee, J. Biomed. Mater. Res., Part A, 2014, 102, 456–469 CrossRef PubMed.
- Y. Liu, Q. Yang, J. H. Wei, R. Xiong, C. X. Pan and J. Shi, Mater. Chem. Phys., 2011, 129, 654–659 CrossRef CAS.
- C. S. Ciobanu, F. Massuyeau, L. V. Constantin and D. Predoi, Nanoscale Res. Lett., 2011, 6, 613 CrossRef PubMed.
- F. Chena, P. Huang, Y. J. Zhua, J. Wua and D. X. Cui, Biomaterials, 2012, 33, 6447–6455 CrossRef PubMed.
- W. Song, Q. Wang, C. Wan, T. Shi, D. Markel, R. Blaiser and W. Ren, J. Biomed. Mater. Res., Part B, 2011, 98, 255–261 CrossRef PubMed.
- A. Mocanu, G. Furtos, A. Danisteanu, G. Rapuntean, C. Prejmerean and M. T. Cotisel, Appl. Surf. Sci., 2014, 298, 225–235 CrossRef CAS.
- S. R. Rout, B. Behera, T. K. Maiti and S. Mohapatra, Dalton Trans., 2012, 10777–10783 RSC.
- Y. P. Guo, L. H. Guo, Y. Yao, C. Q. Ning and Y. J. Guo, Chem. Commun., 2011, 47, 12215–12217 RSC.
- C. Huang, Y. Zhou, Z. Tang, X. Guo, Z. Qian and S. Zhou, Dalton Trans., 2011, 5026–5031 RSC.
- D. F. Mercado, G. Magnacca, M. Malandrino, A. Rubert, E. Montoneri, L. Celi, A. B. Prevot and M. C. Gonzalez, ACS Appl. Mater. Interfaces, 2014, 6, 3937–3946 CAS.
- H. C. Wu, T. W. Wang, M. C. Bohn, F. H. Lin and M. Spector, Adv. Funct. Mater., 2010, 20, 67–77 CrossRef CAS.
- K. Lin, L. Chen, P. Liu, Z. Zou, M. Zhang, Y. Shen, Y. Qiao, X. Liu and J. Chang, CrystEngComm, 2013, 15, 2999–3008 RSC.
- F. Chen, C. Li, Y. J. Zhu, X. Y. Zhao, B. Q. Lu and J. Wu, Biomater. Sci., 2013, 1, 1074–1081 RSC.
- S. Ge, X. Shi, K. Sun, L. Changpeng, C. Uher, J. R. Baker, J. Mark, M. B. Holl and B. G. Orr, J. Phys. Chem. C, 2009, 113, 13593–13599 CAS.
- C. B. Andujar, L. D. Tung and N. T. K. Thanha, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2010, 106, 553–568 RSC.
- A. Demorti, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. B. Colin, Nanoscale, 2011, 3, 225–232 RSC.
- H. Ho, C. P. Tsai, C. C. Chung, C. Y. Tsai, F. R. Chen, H. J. Lin and C. H. Lai, Chem. Mater., 2011, 23, 1753–1760 CrossRef.
- C. A. Demarchi, A. Debrassi, F. C. Buzzi, R. Corrêa, V. C. Filho, C. A. Rodrigues, N. Nedelko, P. Demchenko, A. S. Waniewska, P. Dłużewski and J. M. Greneche, Soft Matter, 2014, 10, 3441–3450 RSC.
- X. Luo, S. Liu, J. Zhou and L. Zhang, J. Mater. Chem., 2009, 19, 3538–3545 RSC.
- Z. Shan, L. Xianghai, Y. Gao, X. Wang, C. Li and Q. Wu, Anal. Biochem., 2012, 425, 125–127 CrossRef CAS PubMed.
- E. B. Ansar, M. Ajeesh, Y. Yokogawa, W. Wunderlich and H. Varma, J. Am. Ceram. Soc., 2012, 95, 2695–2699 CrossRef CAS.
- X. Guo, L. Yu, L. Chen, H. Zhang, L. Peng, X. Guo and W. Ding, J. Mater. Chem. B, 2014, 2, 1760–1763 RSC.
- W. Chen, T. Long, Y. J. Guo, Z. A. Zhu and Y. P. Guo, J. Mater. Chem. B, 2014, 2, 1653–1660 RSC.
- Y. P. Guo, T. Long, S. Tang, Y. J. Guo and Z. A. Zhu, J. Mater. Chem. B, 2014, 2, 2899–2909 RSC.
- K. Lin, L. Xia, J. Gan, Z. Zhang, H. Chen, X. Jiang and J. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 8008–8017 CAS.
- M. Salarian, M. Solati-Hashjin, S. Sara Shafiei, A. Goudarzi, R. Salarian and A. Nemati, Mater. Sci., 2009, 27, 961–971 CAS.
- X. Y. Zhao, Y. J. Zhu, F. Chen, B. Q. Lu and J. Wu, CrystEngComm, 2013, 15, 206–212 RSC.
- C. Qi, Y. J. Zhu, B. Q. Lu, X. Y. Zhao, J. Zhao and F. Chen, J. Mater. Chem., 2012, 22, 22642–22650 RSC.
- R. Gao, X. Mu, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 783–792 RSC.
- C. Qi, Y. J. Zhu, B. Q. Lu, X. Y. Zhao, J. Zhao, F. Chen and J. Wu, Chem.–Eur. J., 2013, 19, 5332–5341 CrossRef CAS PubMed.
- T. Y. Guo, Y. Q. Xia, G. J. Hao, M. D. Song and B. H. Zhang, Biomaterials, 2004, 25, 5905–5912 CrossRef CAS PubMed.
- S. Chukhrai, L. F. Atyaksheva and O. S. Pilipenko, Russ. J. Phys. Chem. A, 2011, 85, 890–896 CrossRef.
- R. Gao, X. Mu, Y. Hao, L. Zhang, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 1733–1741 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07318e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.