
Fabrication of high strength PVA/rGO composite fibers by gel spinning
Jingjing Liab,
Leishan Shaoa,
Xiaohai Zhouc and
Yinghan Wang*a
aState Key Laboratory of Polymer Materials Engineering, College of Polymer Science and Engineering, Sichuan University, Chengdu 610065, PR China. E-mail: wang_yh@scu.edu.cn; Fax: +86-028-85460823; Tel: +86-028-85460823
bMaoming Branch R&D Institute, SINOPEC, Maoming 525011, China
cChengdu Terong New Material Co., Ltd., Chengdu 610502, PR China
First published on 4th September 2014
Abstract
High strength composite fibers were prepared from poly(vinyl alcohol) (PVA) (degree of polymerization: 6100) reinforced by reduced graphene oxide (rGO). The macroscopically homogeneous PVA/rGO dispersion was obtained through solvothermal reduction of graphene oxide (GO) in PVA/dimethyl sulfoxide (DMSO)/H2O solution, and then extruded into composite fibers by gel spinning followed by hot drawing. It was found that the mechanical properties of PVA fibers were greatly improved by incorporating rGO. At 0.1 wt% rGO loading, tensile strength increased from 1.8 GPa for the pure PVA fiber to 2.2 GPa for the PVA/rGO composite fiber. The results of mechanical properties and FTIR spectra for PVA/rGO composite fibers suggest the relatively strong interfacial interactions between rGO nanosheets and PVA that improve the load transfer from the polymer matrix to the reinforcing phase. Meanwhile, the thermal stability of the composite fibers was also enhanced by rGO addition.
1. Introduction
Poly(vinyl alcohol) (PVA) is a flexible-chain polymer with hydroxyl on its side groups, and has good fiber-forming, highly hydrophilic, and good mechanical properties. Thus, it is widely used to fabricate fibers, films for food and drug packages, and barrier membranes, etc.1 Similar to polyethylene, the PVA chain structure in the crystal is planar zigzag. It has been estimated that with perfect alignment (full-extension) of the polymer chains the lattice modulus of PVA could reach 250–300 GPa being calculated theoretical,2–4 which has the potential to produce a high modulus and high strength fiber. However, in spite of various attempts for polymerization control, spinning methods and drawing, the actual tensile strength and modulus of PVA fibers still have a large gap compared with the theoretical value.5To reduce this gap and improve the mechanical properties of PVA fiber, high molecular weight, and high degree of orientation and chain-extension are needed. As is known, the chain terminals of PVA cannot efficiently transfer load, which easily causes stress concentration and then inevitably impairs the mechanical properties. That is to say, the higher molecular weight, the fewer terminal, and higher mechanical properties of PVA fibers. On the other hand, the spinning method is also an important factor for preparing high-performance PVA fibers. PVA fibers could be produced using wet spinning, dry-jet wet spinning, gel spinning, dry spinning, and melt spinning methods, etc.6 Of all these methods, gel spinning is more effective in preparing high-performance fibers than others.7–9 This is because gel spinning allows the gel fiber to be drawn to high extensions, which results in fewer chain entanglements and high degree of chain alignment.4,10
Recently, carbon nanotubes (CNTs) have attracted significant attention due to their unusual physical and mechanical properties such as high tensile strength and high Young's modulus. The incorporation of CNTs into PVA matrix to form composite fibers has attracted much attention because the mechanical properties of PVA fibers could be greatly improved.11–14 Compared with CNTs, as well as its outstanding mechanical, electrical and thermal properties, graphene has also attracted considerable attention because of its low cost and high yield. The incorporation of graphene can dramatically enhance the electrical, mechanical, and thermal properties of polymer composites at extremely low loadings, which result in it being one of the most popular candidates for manufacturing high-performance PVA fibers. Indeed, a few attempts have been made to fabricate PVA/graphene composite nanofibers by electrospinning. For example, Wang et al.15 have fabricated PVA/graphene oxide (GO) composite nanofibers successfully by electrospinning, and found that a very small loading of 0.02 wt% GO increased the tensile strength of the nanofibers by 42 times. Tan et al.16 investigated the effects of nanosheets loading and reduction time of reduced graphene oxide (rGO) on the rheology of GO/rGO nanosheets suspended in PVA solution. It was found that the addition of 1 wt% GO/rGO significantly improved PVA fibrous uniformity and fineness, and the spinnable concentration range of PVA was greatly broadened. Qi et al.17 fabricated PVA/GO nanofibrous biocomposite scaffolds by electrospinning. Efficient load transfer was found between the nanofiller and PVA, and the mechanical properties of PVA/GO composite nanofibers with uniformly dispersion were significantly improved. However, such efforts are very scarce for electrospun PVA/graphene nanofibers, which were not suitable for industrial application to produce high-performance PVA fibers. So it is necessary to introduce graphene into gel-spun PVA fibers to fabricate high-performance fibers.
In this work, we present preparation of PVA/rGO composite fibers through gel spinning to enhance the mechanical properties of PVA fiber by introduction of a small amount of rGO obtained by solvothermal reduction of GO colloidal dispersions in PVA matrix. DMSO/H2O is selected as solvent not only because it is helpful to produce high strength PVA fibers,6,18–20 but because it is a good solvent for GO. More importantly, DMSO is able to reduce GO to rGO.21,22 The PVA/rGO composite fibers have been characterized in terms of their morphological, mechanical, and thermal properties in order to study the effects of rGO nanosheets on their dispersion in the PVA matrix and the interaction with the PVA.
2. Experimental details
2.1 Materials
PVA with an average polymerization degree of 6100 (98–99% hydrolyzed) was supplied by Chengdu Terong New Material Co., Ltd., China. Graphite powder (325 mesh) was purchased from Nanjing XF Nano Material Tech Co., Ltd., China. Potassium permanganate (KMnO4), sulfuric acid (H2SO4, 98%), hydrogen peroxide (H2O2), sodium nitrate (NaNO3), methanol and dimethyl sulfoxide (DMSO) were purchased from Tianjin Bodi Co., Ltd., China. The materials were directly used without further purification. All the water used was deionized.2.2 Preparation of PVA/rGO spinning dope
GO was prepared according to Hummers method as described in detail elsewhere.23,24 The well-dispersed PVA/rGO spinning dope was prepared as follows: GO was dispersed in a DMSO/H2O (92/8 wt%) solution and sonicated (Fisher Scientific bath sonicator, frequency 43 kHz, power 150 W) for 1 h at 25 °C. To obtain PVA/GO mixture, PVA powder was slowly added into GO/DMSO/H2O dispersion, and the mixture was stirred with mild rotation at 50 °C for 20 h, 75 °C for 1 h, 95 °C for 1 h, 105 °C for 3 h and 115 °C for 1.5 h, respectively, then deaerated in an oven at 95 °C for 20 h prior to spinning. The weight contents of GO in the PVA/rGO spinning dopes described above were controlled at three GO contents, namely 0.05, 0.1 and 0.5 wt% relative to PVA content.On the other hand, in order to investigate the reduction degree of GO in PVA spinning dopes, the GO/DMSO/H2O dispersion was heated by the same procedure for preparing PVA/rGO spinning dopes. Then rGO was obtained after filtered and washed with ethanol, finally dried at 40 °C for 24 h in a vacuum oven.
2.3 Gel spinning and hot drawing
The prepared PVA/rGO spinning dopes were transferred to the spinning cylinder under high-purity nitrogen atmosphere. Gel spinning was performed by extruding the solution from a nozzle having a hole size of 0.2 mm (spinneret temperature: 98 °C, nitrogen pressure: 1.3 MPa). The solution was first extruded into open air (air gap, 300 mm), and then immediately coagulated in methanol kept at −17 °C to obtain undrawn PVA gel fibers. The temperature of coagulation bath could be controlled with ±5 °C. Gel fibers were kept rotating in methanol at 30 °C for 5 min and followed by immersing in methanol bath for 30 min at room temperature, subsequently dried at room temperature for 12 h. Then the dried fibers were drawn in an air bath twice at 180 °C (first) and 230 °C (second). An example of a several hundred meters long PVA/rGO composite fiber on the wheels is shown in Fig. 1. | ||
| Fig. 1 Picture of a several hundred meters long PVA/rGO composite fiber on the wheels. | ||
2.4 Characterization
Fourier transform infrared spectra (FTIR) characterizations were performed at ambient temperature with a spectrometer (Nicolet 6700), which is equipped with diffuse reflectance accessories. Wide-angle X-ray diffraction (XRD) pattern of the samples were obtained with X' Pert Pro X-ray diffractometer with Cu Kα radiation (λ = 0.15418 nm) under a voltage of 40 kV and a current of 40 mA. Samples were scanned over the range of diffraction angle 2θ = 2–50°, with a scan speed of 0.5° min−1 at room temperature. Differential scanning calorimetric (DSC) measurements were carried out under dry nitrogen using a DSC analyzer (Netzsch DSC 204 F1) from 40 to 270 °C at 10 °C min−1. Microscopic morphology observations were conducted on a JSM-7500 F scanning electron microscopy (SEM) under an acceleration voltage of 5 kV. The samples were fractured in liquid nitrogen and gold-sputtered prior to observation. Single fiber tensile strength and modulus were measured with A&D Tensilon RTC-1250A according to ASTM standard D3822-7 using a 50 N load cell, gauge length 20 mm and crosshead speed 20 mm min−1. The experimental results were evaluated as the averages of at least 10 measurements. The thermogravimetric analysis (TGA) was performed in a TA-2000 thermo-analyzer instrument (TA Co., USA). The samples were dried under vacuum before the experiments and then placed in a platinum pan. The loss of weight was monitored from 40 to 500 °C at a heating rate of 10 °C min−1 under a nitrogen flow.3. Results and discussion
3.1 rGO characterization
It was reported that DMSO is able to deoxidize GO to certain extent under thermal condition.21,22 In order to determine the extent of GO deoxidation, FTIR was employed to analyze the existence of functional groups and their changes during the process of fabricating PVA spinning dopes. In Fig. 2(a), the pristine graphite only showed two peaks with absorptions at ∼3420 and ∼1630 cm−1, corresponding to the O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, respectively. After the chemical oxidation and exfoliation, the presence of different oxygen-containing groups on the GO surface was confirmed by peaks at ∼3420, ∼1720, ∼1230 and ∼1060 cm−1, which correspond to the stretching vibrations of O–H, CO, C–OH and C–O bonds, respectively.25–27 After solvothermal treatment of GO colloidal dispersions, the partial reduction of GO to rGO is conclusive, based on the significant attenuation of O–H bond peak intensity at 3420 cm−1 and the disappearance of CO bond peak at 1720 cm−1. However, other peaks from C–OH and C–O bonds remain almost unchanged, which indicates that the obtained rGO still remains some oxygenated functionalities. Advantageously, the residual oxygenated functionalities can generate strong interactions with the PVA matrix mainly by hydrogen bonds.28–30
C bonds, respectively. After the chemical oxidation and exfoliation, the presence of different oxygen-containing groups on the GO surface was confirmed by peaks at ∼3420, ∼1720, ∼1230 and ∼1060 cm−1, which correspond to the stretching vibrations of O–H, CO, C–OH and C–O bonds, respectively.25–27 After solvothermal treatment of GO colloidal dispersions, the partial reduction of GO to rGO is conclusive, based on the significant attenuation of O–H bond peak intensity at 3420 cm−1 and the disappearance of CO bond peak at 1720 cm−1. However, other peaks from C–OH and C–O bonds remain almost unchanged, which indicates that the obtained rGO still remains some oxygenated functionalities. Advantageously, the residual oxygenated functionalities can generate strong interactions with the PVA matrix mainly by hydrogen bonds.28–30
 | ||
| Fig. 2 FTIR spectra (a) and XRD patterns (b) of graphite, GO and rGO. | ||
XRD is another widely used characterization method to confirm the structure of GO deoxidation. Fig. 2(b) shows the XRD patterns of flake graphite, GO and rGO. The (001) diffraction peak of flake graphite is located at 2θ = 26.4°, corresponding to d-spacing of 0.34 nm, but it disappears in the pattern of GO. The typical diffraction peak of GO is observed at about 2θ = 10.4°, indicating an increase of interlayer spacing from 0.34 to 0.85 nm due to the intercalating oxide functional groups.31–33 After solvothermal treatment, partially reduced GO shows two peaks, one is at a position close to the typical diffraction peak of GO (2θ = 10.4°), which is attributed to the diffraction of un-reduced GO, and the other belonging to rGO structure appears at 23.8°. Simultaneously, the peak assigned to the GO structure shifts to 12.3°, corresponding to d-spacing of 0.72 nm, suggesting a decrease of oxygen groups in partially reduced GO surface. These results further confirmed that the solvothermal treatment process would reduce GO to some extent, but the reduce extent is not high.
3.2 Dispersion of rGO
Fig. 3 shows photographs and an optical microscope image of GO/DMSO/H2O and PVA/rGO/DMSO/H2O dispersion. All the dispersions were prepared according to the Experimental procedure section. It could be found that GO was well-dispersed in the spinning solvent with brown color (Fig. 3(a)). After solvothermal treatment of the dispersion containing 0.5 wt% GO and 11.8 wt% PVA, the color of the dispersion changed to black (Fig. 3(b)), indicating a successful reduction of GO in the PVA. And the optical microscope image of PVA/rGO/DMSO/H2O dispersion further showed that rGO sheets obtained by solvothermal reduction could disperse homogeneously at the macroscopic level in PVA matrix and no obvious agglomerate could be observed, suggesting that PVA/rGO solution was successfully prepared for gel spinning. | ||
| Fig. 3 (a) DMSO/H2O/GO (0.5 wt%) dispersion, (b) PVA/DMSO/H2O/rGO (0.5 wt%), and (b′) optical micrograph of (b). In both (b) and (b′) the PVA concentration is 11.8 wt%. | ||
XRD is also used to determine whether rGO nanosheets are indeed present as individual graphene sheets in the composite fibers.34 Fig. 4 shows the XRD patterns of PVA fiber and PVA/rGO composite fibers. As for PVA fiber, the diffraction peaks at 2θ of 11.4°, 19.6° and 22.5° corresponded to the crystalline region of PVA.35 As for composite fibers, the XRD pattern showed the same diffraction peak of PVA, while the broad peak of rGO at 2θ = 23.6° disappeared. These results suggested that the rGO sheets were exfoliated and well dispersed into the PVA, which caused the disorder and loss of structure regularity of the graphene. On the other hand, as increasing the amount of rGO, the intensity of the diffraction peak of PVA/rGO composite fibers decreased, which suggested that the crystallinity decreased with increasing rGO loadings. Simultaneously, the crystallinity of PVA composites is further demonstrated by DSC measurement, as shown in Fig. 5. The decrease in crystallinity may be attributed to the “hydrogen bond barrier” effect of rGO nanosheets.28 The fully exfoliated and well-dispersed rGO nanosheets form the “hydrogen bond barrier” among the PVA chains, a physical barrier, which could dramatically diminish the crystallinity of PVA matrix.
 | ||
| Fig. 4 The XRD curves of PVA fiber and PVA/rGO composite fibers. | ||
 | ||
| Fig. 5 The DSC curves of PVA fiber and PVA/rGO composite fibers. | ||
To further evaluate the dispersion state of rGO sheets in PVA and morphology of the composite fibers, the surface and fracture images of PVA fiber and PVA/rGO composite fibers before hot drawing process were characterized by SEM, as shown in Fig. 6. It can be clearly seen that the pure PVA fiber is smooth in surface and stripy in fracture. And the surface and fracture images of PVA/0.1 wt% rGO composite fiber showed a similar structure to pure PVA fiber, which indicated rGO could be well dispersed in the PVA matrix. However, when the amount of rGO increases to 0.5 wt%, the surface of composite fiber gets rougher and the fracture could observe the aggregation of rGO. This phenomenon suggested that, at a high loading level, the rGO nanosheets could not disperse homogeneously at the macroscopic level in PVA matrix, which may limit the reinforcement in the mechanical properties of composite fibers.
 | ||
| Fig. 6 SEM images of surface and cross section of fibers, (a and a2) PVA fiber; (b and b2) PVA-0.1 wt% rGO fiber; (c and c2) PVA-0.5 wt% rGO fiber; (a2 and c2) high magnification SEM images of cross section of fibers. | ||
The interfacial interaction between nanofiller and polymer matrix is a key factor to obtain satisfied composites. The strong interfacial interaction can efficiently transfer the external load and maximize the performance of composite fibers.36 The interaction between rGO nanosheets and PVA chains can be investigated by measuring the extent of shift in FTIR spectrum. Fig. 7 shows the typical band of PVA and PVA/rGO composite fibers. The broad and intense band peaked at 3260 cm−1 is attributed to the symmetrical stretching vibration of hydroxyl group (–OH). The peaks at 2800–3000 cm−1 are due to –CH2 and the peak at 1090 cm−1 results from the stretching of C–O groups. The change of –OH stretching bands is related to hydrogen bonding.28,30,37 From Fig. 7, it can be found that the peak for –OH stretching shifts to a lower wavenumber with the increase of rGO loadings; the band corresponding to –C–OH stretching (around 1095 cm−1) displays a similar behavior. The results indicate that there are strong molecular interactions between rGO nanosheets and PVA chains. The strong molecular interactions at the filler-matrix interface are mainly attributed to the hydrogen bonding between the oxygenated groups remaining in rGO surface and the –OH groups of PVA chains.38
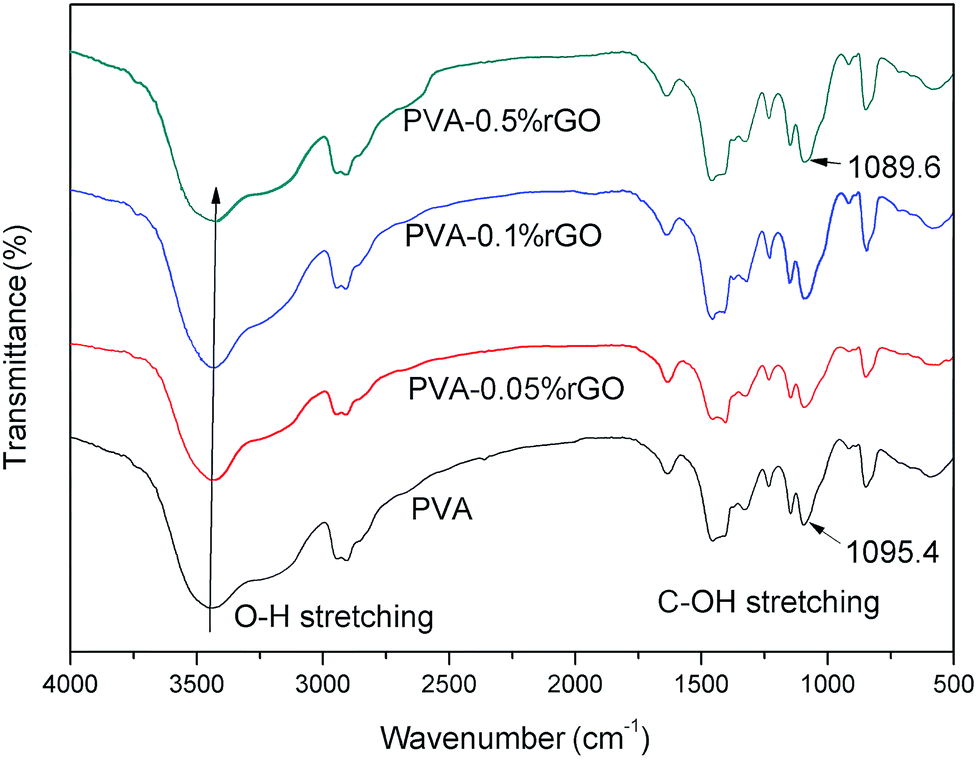 | ||
| Fig. 7 FTIR spectra of pure PVA and PVA/rGO fibers with different rGO loadings. | ||
3.3 Mechanical properties
The general purpose of incorporating a small amount of rGO nanosheets into PVA fiber is to improve its mechanical properties. The tensile properties of pure PVA and PVA/rGO composite fibers are given in Table 1. It is obvious that the addition of rGO into the polymer matrix had a significant influence on the mechanical behavior. With increasing rGO nanosheets, the tensile strength and Young's modulus of PVA/rGO composite fibers obviously increase while the elongation to failure slightly decreases. The maximum tensile strength (2.2 GPa) and Young's modulus (36.5 GPa) are obtained when rGO nanosheets loading is 0.1 wt%. Compared to pure PVA fiber, improvement in tensile strength and Young's modulus by 20.6% and 20.8%, respectively, is observed. Since PVA is a semicrystalline polymer, its mechanical properties strongly depend on the degree of its crystallinity. But according to the results of XRD and DSC (Fig. 4 and 5), a decrease of PVA crystallinity is observed in PVA composite fibers. So the increased strength of the composite fibers cannot be attributed to changes in crystallinity. The reason might be attributed to the homogeneous dispersion of rGO nanosheets in the polymer matrix and strong interfacial interactions between both components. However, when rGO nanosheets addition goes up to 0.5 wt%, the values of their tensile strength and Young's modulus are even lower than that of pure PVA fiber. The reason is that when rGO loadings increased above a critical point, the rGO nanosheets tend to aggregate, which decrease the tensile strength.39| Sample | Draw ratio | Tensile strength (GPa) | Young's modulus (GPa) | Tensile strain (%) |
|---|---|---|---|---|
| PVA fiber | 13.3 | 1.82 ± 0.1 | 30.2 ± 2.4 | 6.5 ± 1.2 |
| PVA-0.05% rGO fiber | 13.1 | 1.98 ± 0.1 | 32.9 ± 2.6 | 6.1 ± 0.8 |
| PVA-0.1% rGO fiber | 13 | 2.20 ± 0.1 | 36.5 ± 3.0 | 5.7 ± 1.5 |
| PVA-0.5% rGO fiber | 11 | 1.60 ± 0.2 | 26.6 ± 3.0 | 5.2 ± 2.0 |
3.4 Thermal properties of PVA/rGO composite fibers
The thermal stability of pure PVA and PVA/rGO composite fibers is investigated. Fig. 8 shows thermogravimetric analysis (TGA) and differential thermogravimetric analysis (DTG) curves of the fibers. Both pure PVA fiber and PVA/rGO composite fibers have the similar decompose curves, but TGA curves of PVA/rGO composite fibers were shifted to a higher temperature compared to that of pure PVA fiber. The onset temperature of degradation for the PVA/rGO composite fibers was higher than that for PVA fiber. The temperature of the maximum degradation rate for the PVA/rGO composite fibers with 0.5 wt% rGO loadings was 367 °C (obtained from the derivative of TGA curves), being 13 °C higher than that of pure PVA fiber. The significantly improved thermal stability of the composite fibers might be attributed to the physical barrier effect of graphene which slowed down the diffusion of pyrolysis products, just like other layered materials such as clay and layered double hydroxides.28 | ||
| Fig. 8 Thermogravimetric curves of PVA and PVA/rGO composite fibers (a) weight loss percent and (b) derivative thermogravimetry curves (DTG). | ||
4. Conclusions
This paper described the preparation of homogeneous PVA/rGO dispersions by solvothermal reduction using DMSO/H2O mixed solvent. The PVA/rGO dispersions were successfully spun into smooth-surfaced composite fibers through gel spinning. The effect of rGO on the morphology, microstructure, mechanical properties and thermal stability of PVA/rGO composite fibers was investigated in detail. A 20.6% increase in tensile strength and 20.8% improvement in Young's modulus have been obtained with only the addition of 0.1 wt% rGO. The increased mechanical strengths of the PVA fiber by the addition of rGO are attributed to the relatively strong interfacial interactions between rGO nanosheets and PVA. The thermal stability of PVA/rGO composite fibers increases obviously with increasing of rGO loadings. From the above, the incorporation of rGO into PVA is an effective method for preparing high-performance PVA fiber. In addition, the processing conditions such as the concentration of PVA and drawing technique can further be optimized.Acknowledgements
This work was supported by National Natural Science Foundation of China [grant number 51173115] and the Ministry of Education [the Foundation for PhD training, grant number 20110181110030] of China. We would like to show our great thanks to Wang Hui (Analytical & Testing Center, Sichuan University) due to her help in SEM observation.References
- W. S. Lyoo, S. S. Han, J. H. Kim, W. S. Yoon, C. J. Lee, I. C. Kwon, J. Lee, B. C. Ji and M. H. Han, Angew. Makromol. Chem., 1999, 271, 46–52 CrossRef CAS.
- I. Satcurada, T. Ito and K. Nakamae, J. Polym. Sci., Polym. Symp., 1967, 15, 75–91 CrossRef PubMed.
- K. Tashiro, M. Kobayashi and H. Tadokoro, Macromolecules, 1977, 10, 731–736 CrossRef CAS.
- M. L. Minus, H. G. Chae and S. Kumar, Macromol. Chem. Phys., 2009, 210, 1799–1808 CrossRef CAS PubMed.
- J. G. Williams, J. Am. Chem. Soc., 1998, 120, 6848–6849 CrossRef CAS.
- W.-I. Cha, S.-H. Hyon and Y. Ikada, J. Polym. Sci., Polym. Phys. Ed., 1994, 32, 297–304 CrossRef CAS PubMed.
- P. Smith and P. Lemstra, J. Mater. Sci., 1980, 15, 505–514 CrossRef CAS.
- P. Smith and P. J. Lemstra, J. Polym. Sci., Polym. Phys. Ed., 1981, 19, 1007–1009 CrossRef CAS PubMed.
- P. Smith, P. Lemstra, B. Kalb and A. Pennings, Polym. Bull., 1979, 1, 733–736 CrossRef CAS.
- H. G. Chae and S. Kumar, Science, 2008, 319, 908–909 CrossRef CAS PubMed.
- X. Xu, A. J. Uddin, K. Aoki, Y. Gotoh, T. Saito and M. Yumura, Carbon, 2010, 48, 1977–1984 CrossRef CAS PubMed.
- X. Zhang, T. Liu, T. V. Sreekumar, S. Kumar, X. Hu and K. Smith, Polymer, 2004, 45, 8801–8807 CrossRef CAS PubMed.
- Z. Wang, P. Ciselli and T. Peijs, Nanotechnology, 2007, 18, 455709 CrossRef.
- C. Mercader, V. Denis-Lutard, S. Jestin, M. Maugey, A. Derre, C. Zakri and P. Poulin, J. Appl. Polym. Sci., 2012, 125, E191–E196 CrossRef CAS PubMed.
- C. Wang, Y. Li, G. Ding, X. Xie and M. Jiang, J. Appl. Polym. Sci., 2013, 127, 3026–3032 CrossRef CAS PubMed.
- Y. Q. Tan, Y. H. Song and Q. Zheng, Nanoscale, 2012, 4, 6997–7005 RSC.
- Y. Qi, Z. Tai, D. Sun, J. Chen, H. Ma, X. Yan, B. Liu and Q. Xue, J. Appl. Polym. Sci., 2013, 127, 1885–1894 CrossRef CAS PubMed.
- K. Yamaura and R. Kumakura, J. Appl. Polym. Sci., 2000, 77, 2872–2876 CrossRef CAS.
- M. Suzuki, T. Tanigami, S. Matsuzawa and K. Yamaura, J. Appl. Polym. Sci., 2002, 86, 1970–1977 CrossRef CAS PubMed.
- T. Tanigami, K. Yano, K. Yamaura and S. Matsuzawa, Polymer, 1995, 36, 2941–2946 CrossRef CAS.
- O. C. Compton, B. Jain, D. A. Dikin, A. Abouimrane, K. Amine and S. T. Nguyen, ACS Nano, 2011, 5, 4380–4391 CrossRef CAS PubMed.
- Z. Lin, Y. Yao, Z. Li, Y. Liu, Z. Li and C.-P. Wong, J. Phys. Chem. C, 2010, 114, 14819–14825 CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- J. Li, L. Shao, L. Yuan and Y. Wang, Mater. Des., 2014, 54, 520–525 CrossRef CAS PubMed.
- J. Ou, J. Wang, S. Liu, B. Mu, J. Ren, H. Wang and S. Yang, Langmuir, 2010, 26, 15830–15836 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. Z. Moghaddam, S. Sabury and F. Sharif, RSC Adv., 2014, 4, 8711–8719 RSC.
- C. Bao, Y. Guo, L. Song and Y. Hu, J. Mater. Chem., 2011, 21, 13942–13950 RSC.
- Y. Xu, W. Hong, H. Bai, C. Li and G. Shi, Carbon, 2009, 47, 3538–3543 CrossRef CAS PubMed.
- H. J. Salavagione, G. Martínez and M. A. Gómez, J. Mater. Chem., 2009, 19, 5027–5032 RSC.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS PubMed.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS PubMed.
- J. J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. F. Ma, T. Y. Guo and Y. S. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS PubMed.
- P.-D. Hong and K. Miyasaka, Polymer, 1994, 35, 1369–1374 CrossRef CAS.
- Z. Tang, Y. Lei, B. Guo, L. Zhang and D. Jia, Polymer, 2012, 53, 673–680 CrossRef CAS PubMed.
- L. Shao, J. Li, Y. Zhang, S. Gong, H. Zhang and Y. Wang, J. Mater. Chem. A, 2014, 2, 14173–14180 CAS.
- H. K. Cheng, N. G. Sahoo, Y. P. Tan, Y. Pan, H. Bao, L. Li, S. H. Chan and J. Zhao, ACS Appl. Mater. Interfaces, 2012, 4, 2387–2394 CAS.
- D. Qian, E. C. Dickey, R. Andrews and T. Rantell, Appl. Phys. Lett., 2000, 76, 2868–2870 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |