
Study on the photophysical and electrochemical property and molecular simulation of broadly absorbing and emitting perylene diimide derivatives with large D–π–A structure†
Long Yanga,
Yuyan Yua,
Jin Zhanga,
Yuanqing Songa,
Long Jianga,
Fang Gaob and
Yi Dan*a
aState Key Laboratory of Polymer Materials Engineering of China, Polymer Research Institute, Sichuan University, Chengdu 610065, China. E-mail: danyichenweiwei@163.com; Fax: +86-028-85402465; Tel: +86-28-85407286
bCollege of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China
First published on 27th August 2014
Abstract
To obtain broadly absorbing perylene derivatives with the synchronous impact of the intrinsic π–π* transition of a large conjugated system and intramolecular charge transfer (ICT), we report here the facile synthesis and physical characterization of new N,N′-(di-n-octyl)-3,4,9,10-perylenetetracarboxylic acid diimides (PDI) bearing benzyl-substituted quinoline-4(1H)-ylidene-methyl units as effective donors on the 1,7-positions (C1) and 1-position (C2). Compared to unsubstituted PDI, C1 and C2 both show a pronounced bathochromic shift to the near-infrared region, a high molar extinction coefficient, concentration-dependent π–π-stacking-induced fluorescence and a low LUMO energy level. The investigation of spectroscopic properties and molecular simulation reveal an effective ICT character in compounds C1 and C2. All the properties and analysis indicate that the derivatives are efficient at solar harvesting and are potential candidate materials for use in photovoltaic devices and photocatalysts.
Introduction
To make full use of solar energy in the aspects of renewable energy sources and construction of solar energy conversion systems, many research efforts have focused on the exploration of molecular photoelectric devices in photochemical energy conversion, which requires high light-harvesting and electron-transporting capacity and excellent photo-induced intramolecular charge transfer (ICT). For this purpose, recent developments in the field of organic photovoltaics have aroused great interest in the design of an innovative class of photo-functional polymers and dyes or pigments exhibiting a broad light-harvesting range in the visible and near-infrared (NIR) region to attain more efficient use of solar energy.1 The unavoidable shortcoming of the materials used is their high cost. Accordingly, dyes or polymers with a facile modification synthetic route will be in huge demand. In the meantime, materials with a lower LUMO energy level will facilitate electron transport in hybrid materials2 or from the light-harvesting unit to the conduction band of a semiconductor (i.e. TiO2, ZnO).3Perylene diimides (PDI), a class of industrial dyes which represent highly chemically, photo- and thermostable n-type semiconductors4 with relatively good electron affinity, a high molar extinction coefficient and excellent transport property are potential candidates as electron-accepting materials in many contexts such as organic photovoltaic solar cells,5 light-harvesting arrays,6 organic–polymer hybrid materials7 and photo-induced energy and electron-transfer processes.8 Moreover, high electron mobility via π–π stacking favors ICT and increases the charge separation.9 In this regard, the selection of a suitable donor–π–acceptor (D–π–A) structure with a large conjugated system is the most important parameter for achieving the above goals.
Extending the intrinsic light absorption range (400–550 nm) of PDI,10 substitution in the bay region of PDI through C–C or C–N coupling is a simple route to modify the HOMO–LUMO level, acting to extend the visible-NIR absorption region of the derivatives as compared to unsubstituted PDI.11 The former methods comprised several synthetic steps with high cost, and the light absorption of some derivatives could not cover the whole visible-NIR range. In the meantime, the introduction of a strong electron-donating substituent which enhances the ICT effect unfavorably increases the LUMO energy level,12 and unfortunately a high LUMO level will not favor the charge transfer process.13
Thus, a new idea is put forward: by introducing a conjugated substituent without perturbing the electronic transition of the perylene ring excessively, the synchronous impact of the π–π* transition absorption of the large conjugated system and the ICT transition absorption can broaden the visible-NIR absorption without significantly increasing the LUMO energy level. Moreover, recent studies revealed that the presence of a quinoidal fragment in the molecules of NIR absorbing dyes led to a significant bathochromic shift in their absorption.14 To verify this proposition and obtain visible-NIR-absorbing light-harvesting sensitizers and materials with a large D–π–A structure, under the above guideline target compounds with a merocyanine substituent through a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond will be introduced onto the bay region of the perylene ring with a quinoidal push–pull structure and the diimide moiety as the acceptor. The heavy atom Br will be introduced as a medium electron-withdrawing substituent to demonstrate the effect on the LUMO energy15 and the effect on the aggregation behavior and, for comparison, the strong donor (n-octylamine)-substituted perylene derivative (C3) will also be synthesized and investigated.
C bond will be introduced onto the bay region of the perylene ring with a quinoidal push–pull structure and the diimide moiety as the acceptor. The heavy atom Br will be introduced as a medium electron-withdrawing substituent to demonstrate the effect on the LUMO energy15 and the effect on the aggregation behavior and, for comparison, the strong donor (n-octylamine)-substituted perylene derivative (C3) will also be synthesized and investigated.
Here, a simple synthetic method will be applied to obtain perylene derivatives with a large D–π–A system, light absorption covering the range 400–750 nm and low LUMO energy. In addition to the research on photo-absorption property, the unusual fluorescence emission, electrochemical properties and molecular modeling will also be investigated and discussed.
Experimental section
Starting materials
Perylenetetracarboxylic acid anhydride, liquid bromine, n-octylamine and benzyl bromide were purchased from Chengdu Astatech Trading Co., Ltd. 4-Methylquinoline and N-ethyldiisopropylamine were obtained from Suzhou Yacoo Corporation and Adamas-beta (Shanghai, China), respectively. All the starting materials and reagents were used without further purification. Column chromatography was performed on silica gel (70-80A, Qingdao Hailang).Analysis instruments
1H NMR spectra were recorded on a Bruker Avance II −400 MHz. UV/vis spectra were recorded on a UV-2300 spectrophotometer (Hitachi, Japan). Fluorescence spectra were recorded on an F4600 spectrometer (Hitachi, Japan) excited at 430 nm for solution and 400 nm for thin film. The molecular ground-state geometries were optimized using Becke's three-parameter hybrid exchange–correlation (XC) functional B3LYP16 and 6-31G (d,p) basis which were performed using the Gaussian 03 software package.17 The electrochemical measurements were carried out on the CIMPS workstation (Zahner Elektrik Co., Germany).Synthesis and characterization
The chemical structures of the target compounds and the synthetic routes used for the preparation are outlined in Fig. 1 and Scheme 1. Further synthetic procedures are described as follows.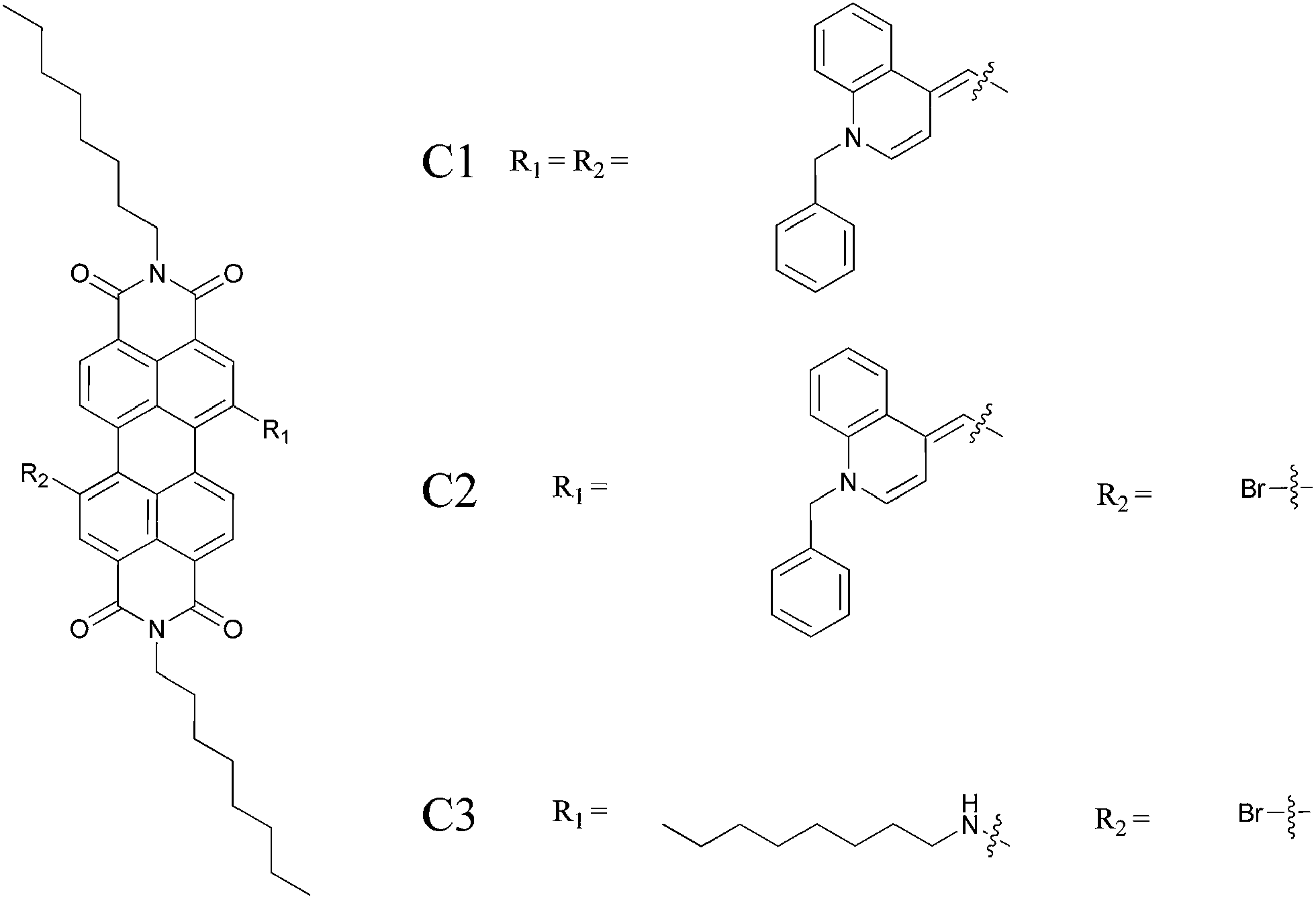 | ||
| Fig. 1 Structures of the compounds C1, C2 and C3. | ||
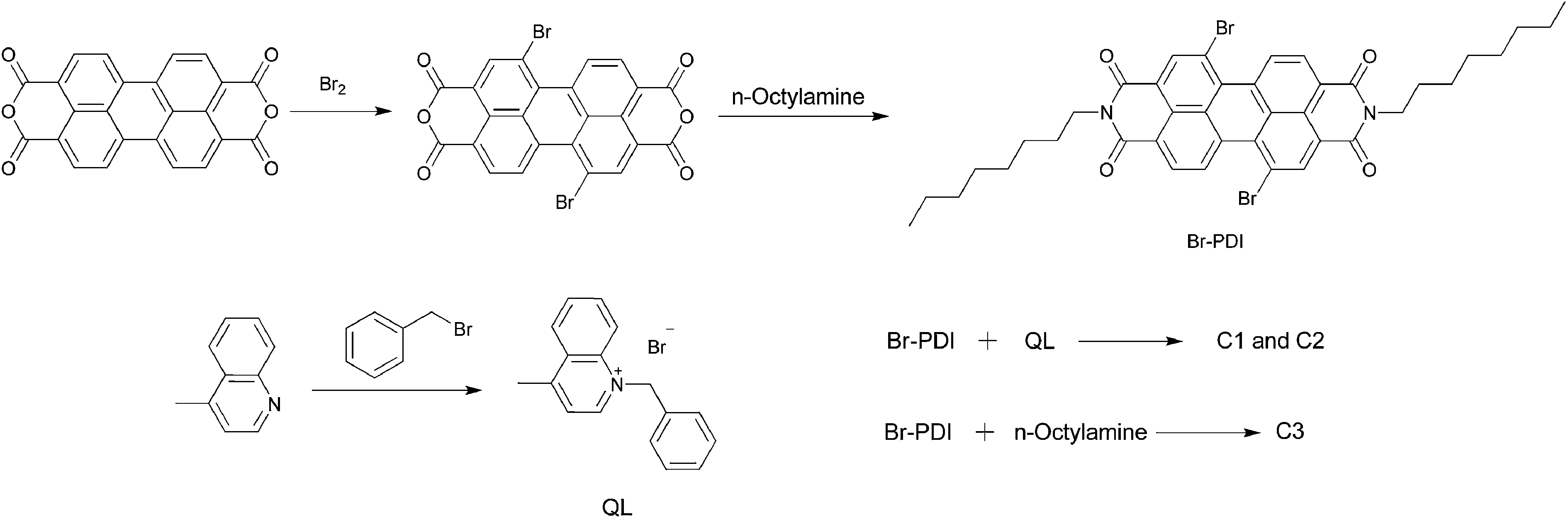 | ||
| Scheme 1 Synthetic routes of C1, C2 and C3. | ||
Br-PDI
Br-PDI was synthesized according to the literature.18 To a solution of concentrated sulfuric acid (150 mL) was added perylenetetracarboxylic acid anhydride (5.0 g, 12.7 mmol). After stirring for 0.5 h at room temperature, the mixture was warmed to 55–60 °C, and iodine (0.3 g, 1.27 mmol) was added. After 3 h, bromine (1.7 mL, 31.8 mmol) was added to the mixture slowly which was then warmed to 85 °C and stirred for 8 h. After bubbling excess bromine with N2 gas into aqueous NaOH solution, the mixture was quenched with ice water, filtered under reduced pressure, washed with water, and dried under vacuum to give a red solid (6.5 g) which was used in the next step directly.A suspension of brominated perylene dianhydrides (5.0 g, 90.9 mmol) obtained in the above reaction, n-octylamine (3.5 g, 27.1 mmol), and acetic acid (2.5 g, 41.7 mmol) in 50 mL N-methyl-2-pyrrolidinone was stirred at 85 °C under Ar for 8 h. After cooling to room temperature, the mixture was poured into water, and the precipitate was separated by filtration, washed with 100 mL MeOH, then dried under vacuum at 40 °C. The crude product was purified by silica gel column chromatography with CH2Cl2–hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. A mixture of 1,7- and 1,6-dibromoperylene diimides was obtained as a red powder. The regioisomeric 1,7-dibromoperylene diimide (3.2 g, yield 42.4%) was obtained through two recrystallization procedures. 1H-NMR (400 MHz, CDCl3): δ = 9.420–9.440 (d, J = 8.0 Hz, 2H), 8.873 (s, 2H), 8.648–8.668 (d, J = 8.0 Hz, 2H), 4.176–4.214 (t, J = 7.6 Hz, 4H), 1.728–1.762 (m, 4H), 1.252–1.438 (m, 20H), 0.865–0.898 (t, J = 6.6 Hz, 6H).
:1) as eluent. A mixture of 1,7- and 1,6-dibromoperylene diimides was obtained as a red powder. The regioisomeric 1,7-dibromoperylene diimide (3.2 g, yield 42.4%) was obtained through two recrystallization procedures. 1H-NMR (400 MHz, CDCl3): δ = 9.420–9.440 (d, J = 8.0 Hz, 2H), 8.873 (s, 2H), 8.648–8.668 (d, J = 8.0 Hz, 2H), 4.176–4.214 (t, J = 7.6 Hz, 4H), 1.728–1.762 (m, 4H), 1.252–1.438 (m, 20H), 0.865–0.898 (t, J = 6.6 Hz, 6H).
QL
The reaction mixture of 4-methylquinoline (5.0 g, 34.5 mmol) and benzyl bromide (11.8 g, 69.0 mmol) was heated to reflux for 3.0 h. After cooling to room temperature, the mixture was poured into absolute ether, and the formed precipitate was separated by filtration, washed and dried under vacuum to give a light gray solid (9.8 g, yield 89.9%) which was used directly in the next step.C1 and C2
C1 and C2 were synthesized according to the literature.19 Br-PDI (0.50 g, 0.65 mmol) and the quaternary intermediate QL (0.41 g, 1.30 mmol) were dissolved in 15 mL NMP in a Schlenk flask and were flushed with argon. The reaction mixture was heated to 115 °C and a mixture of N-ethyldiisopropylamine (0.50 g, 3.90 mmol) and pyridine (0.62 g, 7.80 mmol) was added rapidly. After 45 minutes at this temperature, the reaction mixture was cooled to room temperature and poured into 150 mL of a 3:1 mixture of distilled water–acetone. The formed precipitate was suction-filtered, washed with water, and dried under vacuum. The crude product was further separated through column chromatography on silica gel (methylene chloride–hexane = 3:1) to give C1 (dark purple solid, 0.30 g, yield 43%) and C2 (dark red solid, 0.15 g, yield 25%).
C1, 1H-NMR (400 MHz, CDCl3): δ = 8.501–8.576 (m, 2H), 8.353–8.370 (d, J = 6.8 Hz, 2H), 7.393–7.430 (t, J = 7.4 Hz, 5H), 7.283–7.352 (m, 8H), 7.152–7.229 (m, 1H), 7.033–7.110 (m, 2H), 6.685–6.811 (m, 4H), 4.755–4.838 (m, 6H), 4.038–4.131 (m, 4H), 3.942–3.997 (dd, J = 6.4 Hz, 2H), 3.737–3.794 (dd, J = 6.0 Hz, 2H), 1.660–1.693 (m, 4H), 1.257–1.390 (m, 20H), 0.846–0.863 (m, 6H).
C2, 1H-NMR (400 MHz, CDCl3): δ = 8.547–8.615 (m, 4H), 8.383 (s, 1H), 7.384–7.421 (t, J = 7.4 Hz, 2H), 7.255–7.338 (m, 3H), 7.141–7.159 (d, J = 7.2 Hz, 1H), 7.046–7.085 (t, J = 7.8 Hz, 1H), 6.706–6.743 (t, J = 7.4 Hz, 2H), 6.197–6.216 (d, J = 7.6 Hz, 1H), 4.746–4.824 (m, 3H), 4.105–4.183 (m, 4H), 3.924–3.966 (d, J = 16.8 Hz, 1H), 3.711–3.753 (d, J = 16.8Hz, 1H), 1.693–1.729 (m, 4H), 1.269–1.427 (m, 20H), 0.832–0.869 (m, 6H).
C3
A suspension of Br-PDI (0.20 g, 0.26 mmol), n-octylamine (0.02 g, 0.13 mmol) and pyridine in catalytic amount in NMP (10.0 mL) was vigorously stirred and heated at 70 °C under argon for 1 h. After being cooled to room temperature, the reaction mixture was added to water (50.0 mL) slowly. The resulting green precipitate was filtered off and repeatedly washed with water and methanol. After drying under vacuum, the crude product was purified by column chromatography on silica gel (methylene chloride) to give C3 (0.11 g, yield 87.1%) as a dark green powder. 1H-NMR (400 MHz, CDCl3): δ = 8.660–8.680 (d, J = 8.0 Hz, 1H), 8.432–8.452 (d, J = 8.0 Hz, 1H), 8.317–8.337 (d, J = 8.0 Hz, 1H), 8.200–8.220 (d, J = 8.0 Hz, 1H), 8.114–8.135 (d, J = 8.0 Hz, 1H), 8.026–8.046 (d, J = 8.0 Hz, 1H), 5.932 (s, 1H), 4.137–4.173 (t, J = 7.2 Hz, 4H), 3.426–3.453 (t, J = 5.4 Hz, 2H), 1.835–1.870 (m, 2H), 1.735–1.769 (m, 4H), 1.318–1.622 (m, 30H), 0.87–0.93 (m, 9H).Results and discussion
Photophysical properties – absorption
The steady-state UV-visible spectra of C1, C2 and C3 were recorded in 1.0 × 10−5 M solutions in CHCl3 (Fig. 2). The perylene derivatives all have broad absorption in the visible light range from 400 to 750 nm. C1 and C2 both exhibit typical π–π* transitions in the range 400–580 nm (S0 → S1 transition)20 with a well-resolved vibronic structure that can be attributed to breathing vibrations of the perylene skeleton, whereas the respective breathing vibrations in compound C3 are perturbed by the amine group, which results in a red shift of broad absorption in the 520–750 nm region. The higher energetic S0 → S2 transition is also observed at absorption maxima around 385 nm (for C1 and C2) and 429 nm (for C3).21 Due to the larger conjugation degree (44 aromatic electrons for C1 and 32 aromatic electrons for C2) caused by CC bonds connected to the quinoline moiety, within the 400–580 nm range the absorption maxima appear to be red-shifted for C1 (30 nm) and C2 (20 nm) compared to bay-unsubstituted perylene derivatives.22 Moreover, the target compounds present an obvious long-wavelength absorption with ICT characteristics in the range 580–750 nm (S0 → S1 transition), which confirms effective charge transfer between the donor (merocyanine part) and acceptor (perylene diimide part). Notably, the breathing vibrations of the perylene skeleton in C1 and C2 are not perturbed by the ICT effect and the quinoidal-structure substituent group, which results in broad absorption with a high molar extinction coefficient (Table 1).
 | ||
| Fig. 2 Absorption properties of C1–C3 in CHCl3 solution (1.0 × 10−5 M). | ||
Moreover, UV-visible spectra of C1, C2 and C3 were recorded at different concentrations (0.05 × 10−5 to 10.0 × 10−5 M) in CHCl3 in order to understand the aggregation properties of the perylene diimide derivatives (Fig. 3). In the visible-NIR range, the intensity of both π–π* vibronic transitions and ICT absorption for the whole range of concentrations (0.5 × 10−5 to 5.0 × 10−5 M) do not vary considerably, whereas in the concentrated solution (10.0 × 10−5 M) the absorption peaks and intensity of vibronic transitions both change dramatically. Thus, the aggregation effect can be excluded at concentrations up to 5.0 × 10−5 M according to the photo-absorption property. Consequently, absorption and photoluminescence measurements will not be perturbed by aggregation between molecules in dilute solutions in a measurement range up to this concentration.
 | ||
| Fig. 3 Concentration–dependent absorption properties of C1 (a), C2 (b) and C3 (c) in CHCl3 solution (×10−5 M). | ||
The frontier orbital HOMO–LUMO energy gap (Egap) was determined from the UV-visible absorption spectra23 (Table 1). The Egap values of C1 (1.632 eV) and C2 (1.636 eV) are slightly lower than that of C3 (1.664 eV) due to the much larger degree of conjugation in C1 and C2, which also results in an obvious red shift of the absorption maxima of the perylene skeleton (Fig. 2).
Photophysical properties – emission
To study the excited-state property, fluorescence spectra (Fig. 4) of C1, C2 and C3 were recorded in CHCl3 solution and all the samples were selectively excited at 430 nm (the selection of an excitation wavelength in the visible-light range was based on the exclusion of frequency peak interference with the emission peak) in the same conditions. In Fig. 4b, at a concentration of 5.0 × 10−5 M the derivatives all exhibit broad emission in the range 450–850 nm, among which the 450–650 nm range corresponds to radiation of the excited state of the perylene skeleton, while the 650–850 nm range corresponds to the ICT (S1 → S0 transition) characteristic. Remarkably, the heavy atom (Br) in C2 and C3 does not play a dominant role in the change in emission peaks (Table 2). The emission maxima in the wavelength range 450–650 nm increase in the order C3 → C2 → C1 which can be attributed to the increase in conjugation degree of the perylene ring with a merocyanine substituent. However, the ICT emission peak exhibits an order of C3 > (C1 and C2), mainly because the lone electrons of the amine substituent offer isolated electrons and remarkably enhance ICT between the donor (amine group) and acceptor (diimide moiety) (Fig. 5). | ||
| Fig. 4 Emission property of C1 (a, × 10−5 M), C1–C3 (b, normalized) in CHCl3 solution. | ||
| Compound | λmax (nm) | ICT stokes shift (nm) | |||
|---|---|---|---|---|---|
| C1 | 484 | 532 | 585 | 719 | 92 |
| C2 | 481 | 529 | 576 | 715 | 93 |
| C3 | 481 | 515 | — | 723 | 97 |
 | ||
| Fig. 5 ICT emission spectra of C1–C3 in CHCl3 solution (5.0 × 10−5 mol L−1, normalized). | ||
Notably, the derivative C1 exhibits obvious intensity and peak change in emission (Fig. 4a) in the long-wavelength (650–850 nm) range at different concentrations, which can be attributed to effective charge transfer between the donor (merocyanine part) and acceptor (perylene diimide part) (ICT emission peak). As concretely illustrated in depth in Fig. 6, with the increase in concentration of the dilute solution from 0.6 × 10−5 M to 5.0 × 10−5 M, the intensity slightly increases and the emission maxima show a slight change. When the concentration increases up to 1.0 × 10−4 M, the emission maxima present an apparent red shift, and at a concentration of 5.0 × 10−4 M the intensity decreases and the emission maxima present a dramatic red shift indicating the emergence of intermolecular action which consequently results in the absence of the emission peak in the short-wavelength range (450–650 nm) due to strong π–π interaction of the perylene rings upon aggregation.24 This is a remarkable result, because aggregation often opens up new pathways for quenching of the excitation energy; this can be observed for many extended π systems, including many perylene derivatives in the solid state.25 A similar critical concentration (1.0 × 10−4 M) for apparent intermolecular action can be deduced from fluorescence spectra compared with UV-visible absorption spectra (5.0 × 10−4 M).
 | ||
| Fig. 6 Concentration-dependent ICT emission changes of C1 at different concentrations in CHCl3 (a: × 10−5 M; b: the ICT emission maxima over concentration). | ||
To study systematically the aggregation-related behavior of fluorescence emission, the concentration-dependent intensity changes of each emission peak in the range 450–850 nm are shown in Fig. 7. For the concentration of C1 in CHCl3, the intensities of the emission peaks (W1, W2, W3 and W4) all increase normally until a critical concentration at which the probability of intermolecular action and the effect of fluorescence quenching increase. Notably, upon a concentration increase to 5.0 × 10−4 M, the fluorescence intensity of the ICT emission peak appears higher than that of the short-wavelength emission of the perylene skeleton. Remarkably, the ICT emission peak retains its shape, with a slight shoulder peak on the right, and no new long-wavelength emission is observed, hence emission of an excimer is excluded. The same results have also been obtained for C2 and C3. This phenomenon could be attributed to π–π-stacking-induced quenching of short-wavelength emission and enhancement of ICT-character emission due to the disappearance of short-wavelength emission peaks (W2, W3 and W4).
 | ||
| Fig. 7 Intensity changes of emission peaks of C1–C3 at different concentrations (CHCl3 solution). Inset: marks of the emission peaks of C1. | ||
To investigate further the aggregation-induced photophysical behavior of the derivatives in the solid state, the UV-visible absorption spectra and fluorescence spectra (Fig. 8) were performed on quartz glass on which thin films were obtained through dip-evaporation. C1, C2 and C3 all show broad absorption from 400 nm to 900 nm which makes the derivatives very suitable for light-harvesting materials. The disappearance of a well-resolved vibronic absorption structure can be attributed to the aggregation of ground-state molecules through stacking of the π–π conjugated plane, which was further verified through the disappearance of the 500–650 nm emission peaks in the solid state on quartz glass. The strong π–π interactions also lead to an almost complete loss of fine structure in the fluorescence emission. Interestingly, compared with the fluorescence behavior in CHCl3 solution, the 750 nm ICT emission peak markedly decreases.
 | ||
| Fig. 8 Normalized absorption (a) and emission (b, excitation at 400 nm) properties of C1, C2 and C3 in thin film on quartz glass. | ||
The 800–850 nm emission peak might be assigned to the frequency peak of the excitation light. To gain more information about the aggregation behavior of the derivatives with large π-conjugated systems, the self-assembly property will be investigated in the next research focus.
The Stokes shift is defined as the difference between the absorption and emission maxima which can be used to evaluate the non-radiative energy loss in the excited state. The ICT absorption and emission of the three derivatives all exhibit a large Stokes shift: 92 nm (C1), 93 nm (C2) and 97 nm (C3) (Tables 1, 2 and Fig. 9) corresponding to 2025 cm−1, 2096 cm−1 and 2140 cm−1 in wavenumbers, respectively. The large Stokes shift also indicates the effective ICT characters of C1 and C2 and the non-radiative energy loss which could be partially explained by the large dihedral angles between the acceptor (perylene ring) and the donor (quinoline moiety) in the ground state (see Computational analysis).
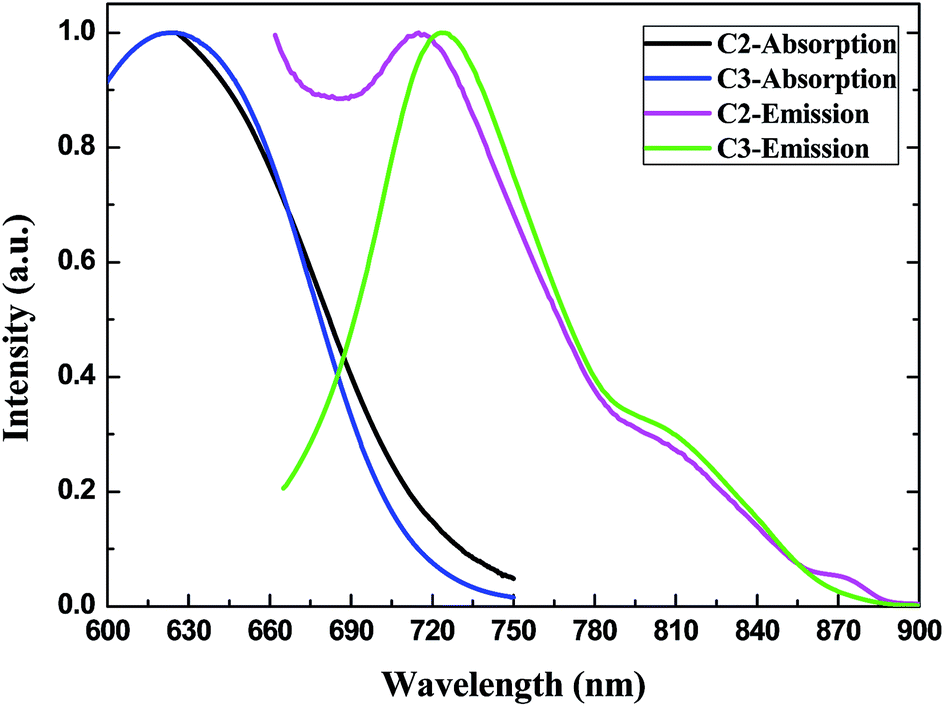 | ||
| Fig. 9 Normalized absorption and emission of C2 and C3 in CHCl3 solution (0.5 × 10−4 M). | ||
Computational analysis
In order to support and clarify the experimental results further, theoretical calculations on the molecules were performed to gain deeper insight into the optimized structure, the electric properties and the electron density distribution of the frontier orbital.The dipole moments (Table 3) calculated by the DFT method in the ground state are 6.79 D (C1), 9.30 D (C2) and 4.73 D (C3). The large values indicate the strongly asymmetric distribution of positive charge and negative charge and the potential of the derivatives. The dipole moments of C1 and C2 are much larger than that of C3, so reasonably the ICT character of C1 and C2 is much stronger. Through geometry optimization, the non-planarity of the perylene core is discovered, and the dihedral angles of the perylene core (18.15° for C1, 18.33° for C2 and 18.31° for C3) (Table 3 and Fig. 10) for C2 and C3 are within the dihedral angle range of common perylene diimide derivatives (0–38°). Remarkably, the small dihedral twist angles of C1, C2 and C3 reveal the concentration-dependent unusual fluorescence induced by molecular aggregation.26 The dihedral angles between the perylene ring and quinoline moiety for C1 (38.05° and 40.76°) and C2 (36.53°), and the amine substituent and perylene ring for C3 (10.78°) are also given.
| Compound | Dipole (D) | HOMO (eV) | LUMO (eV) | Egap (eV) | Dihedral (°) | |
|---|---|---|---|---|---|---|
| D1 | D2 | |||||
| C1 | 6.79 | −4.52 | −2.86 | 1.66 | 18.15 | 38.05/40.76 |
| C2 | 9.30 | −4.91 | −3.14 | 1.77 | 18.33 | 36.53 |
| C3 | 4.73 | −5.54 | −3.25 | 2.29 | 18.31 | 10.78 |
 | ||
| Fig. 10 Schematic diagram of dihedral angles of the derivatives. | ||
The ICT effect can also be elucidated through the electron densities of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). For C1 and C2, the HOMO is mainly located on the quinoline moiety whereas the LUMO is more spread over the perylene region, representing a large degree of intramolecular charge transfer in the ground state. However, for C3 the HOMO and LUMO are both mainly located on the perylene ring.
As clearly shown in Table 3, the calculated HOMO and LUMO energy levels in the ground-state optimized geometry are found to be −4.52 eV and −2.86 eV for C1, −4.91 eV and −3.14 eV for C2 and −5.54 eV and −3.25 eV for C3, respectively, which are in good agreement with the electrochemically determined HOMO and LUMO energy levels (Table 4). Comparing the HOMO energy levels, the increase in the values (−4.52 eV > −4.91 eV > −5.54 eV) could be assigned to the larger degree of π-conjugation spread over the perylene ring and the quinoline moiety and the electron-donating effect of the nitrogen in the quinoline moiety as shown in the electron density distribution of the HOMO. In contrast, the incorporation of an electron-withdrawing substituent (Br) fails to significantly lower the LUMO energy levels. The increase in HOMO energy level and decrease in LUMO energy level effectively lead to a relatively narrow band gap in C1 (1.66 eV), which is in good accordance with the value determined from the λonset of the UV-vis absorption spectra. A conclusion can be drawn that the incorporation of a quinoline substituent through a CC bond can effectively extend the π-conjugated system, enhance the ICT extent, and accordingly narrow the band gap and broaden the photo-absorption range without significantly perturbing the vibronic transition of the perylene skeleton.
To illustrate the π–π stacking induced unusual emission from the point of view of molecular frontier orbitals for the first time, the electronic structures of the frontier orbitals of C1 are also discussed. Fig. 11 and 12 show the electron density distribution and energy levels of the frontier molecular orbitals (HOMO − 2, HOMO − 1, HOMO, LUMO, LUMO + 1 and LUMO + 2) of C1. In the same way as HOMO, the electron density of HOMO − 1 is spread over the whole conjugated system, while HOMO − 2 is predominantly located on the perylene skeleton, respectively. On the other hand, the electron density of LUMO + 1 is located on the perylene ring, while LUMO + 2 is spread mainly over one side of the whole conjugated system. The Egap values of HOMO − 1–LUMO, HOMO − 2–LUMO, HOMO–LUMO + 1 and HOMO–LUMO + 2 are 2.09 eV, 2.80 eV, 3.10 eV and 3.12 eV, respectively. The λonset of the 560 nm absorption peak is about 580 nm (2.13 eV) which is well matched with the Egap of the HOMO − 1–LUMO transition. Thus, supported by the former research,27 we predict that the electronic transition corresponding to the absorption peak of 560 nm mainly originates from the excitation from HOMO − 1 to LUMO. Moreover, the electronic transitions HOMO − 1–LUMO, HOMO − 2–LUMO, HOMO–LUMO + 1 and HOMO–LUMO + 2 all exhibit ICT character to some extent and this behavior might be another factor that causes the red shift of the absorption maxima of the perylene skeleton. Remarkably, as the concentration would have a significant influence on the ICT behavior, the electronic transitions HOMO − 1–LUMO, HOMO − 2–LUMO, HOMO–LUMO + 1 and HOMO–LUMO + 2 with ICT character may be another factor that results in the unusual change in fluorescence emission of the W2–W4 peaks in concentrated solutions as elucidated in the fluorescence analysis.
 | ||
| Fig. 11 Electron density distribution of frontier orbitals of C1–C3. | ||
 | ||
| Fig. 12 Computed molecular orbital energy levels diagram of C1. | ||
Cyclic voltammograms
Further insight into the electronic properties of the perylene derivatives was gained by cyclic voltammetry. The experiments were performed in a three-electrode system using a platinum plate as working electrode and Ag/AgNO3 as reference electrode in acetonitrile solvent containing 0.1 M n-Bu4NPF6 as conducting electrolyte at a scan rate of 100 mV s−1. The samples were dissolved in acetonitrile solution (1.0 × 10−4 M). The solutions were purged with argon for 25 min before each measurement. All potentials were internally referenced to the Fc (ferrocene)/Fc+ couple.28For C1 and C2, the cyclic voltammograms (Table 4 and Fig. 13) show two characteristic oxidation and reduction potentials between −0.7 and −1.4 V, which correspond to the reduction of perylene diimide to dianionic species.30 Concretely, C1, C2 and C3 show irreversible oxidation potentials at 0.49 V, 0.53 V and 0.47 V which imply the oxidation of the substituent moiety (quinoline part for C1 and C2, alkylamine part for C3, respectively) by which the HOMO is mainly contributed, while the reversible redox cycle between −0.7 and −1.4 V represents the electrochemical reaction of the diimide moiety on which the LUMO is mainly located in C1 and C2. Thus the ICT character of the three derivatives is verified through their electrochemical property.
 | ||
| Fig. 13 Cyclic voltammograms of C1, C2 and C3. | ||
The decrease in the electrochemical band gap is obtained through the introduction of electron-donating- and accepting groups into the bay region of PDI. As commonly accepted, the alteration in the HOMO level depends on the oxidation potential of the donating substituent, while the LUMO level is altered according to the reduction potential of the accepting substituent. As the HOMO and LUMO both have a node on the nitrogen atom of the imide, the nitrogen substituent on the diimide part has little effect on the photophysical behavior or the LUMO energy level. Therefore, the change in the HOMO and LUMO energy levels should be attributed to the introduction of a quinoline moiety for C1 and C2. Notably, the LUMO is partially located on the quinoline group (Fig. 11), thus the decrease in the LUMO energy level of C1 and C2 can be well explained. Moreover, the LUMO of C2 (−3.83 eV) is slightly lower than that of C1 (−3.80 eV), thus the introduction of the heavy atom Br has little impact on the LUMO energy level. However, compared with C2 (−5.33 eV), the increase in the HOMO for C1 (−5.29 eV) can be attributed to the improvement in conjugated content, while the increase in the HOMO for C3 (−5.27 eV) can be explained by the introduction of isolated p electrons on the nitrogen atom of the n-octylamine substituent.
For C1 and C2, the electrochemical band gaps (Egap) are calculated as 1.49 eV and 1.50 eV respectively, which are lower than any other PDI derivatives of the same type within our range of knowledge. In the meantime, the band gaps (Egap) calculated through the oxidation and reduction potentials are very close to the values obtained from optical absorption spectra (Table 4) and DFT calculations (Table 3). But for C3, the band gap (2.29 eV) calculated through the molecular model is higher than the values obtained from optical absorption spectra (1.66 eV) and cyclic voltammetry (1.56 eV).
It is believed that the suitable HOMO and LUMO energy levels of the organic–polymer hybrid material would be −5.4 eV and −3.9 eV respectively.31 Moreover, the electron affinity with a LUMO energy level around −3.8 eV (similar to the LUMO level of widely used PCBM32 and the polymer acceptor on alternating dithienothiophene and perylene diimide units33) can facilitate the electron acceptor for use in a polymer bulk heterojunction device. Accordingly, C1 and C2 are promising materials for replacing PCBM when used for further increasing photovoltaic performance, together with their excellent characteristics of broad solar light absorption and ICT. Moreover, when used as a donor for efficient bulk heterojunction solar cells utilizing PCBM as the electron acceptor, the energy levels of C1 and C2 fit very well with the required energy levels (EHOMO level between −5.2 and −5.8 eV; ELUMO level between −3.8 and −4.0 eV).34 Taking into account a LUMO energy level for PCBM at −4.3 eV and using a semi-empirical estimation equation,35 the calculated open-circuit voltages (Voc) are ca. 0.69 V for C1 and 0.73 V for C2, respectively, which are close to the data obtained from a poly(2,7-carbazole-DPP) derivative as the donor.36
Conclusion
In summary, merocyanine-substituted D–π–A structural perylene derivatives with a large π-conjugated system (44 aromatic electrons for C1) were synthesized through a simple synthetic method and investigated using UV-visible and fluorescence spectroscopy, cyclic voltammetry and geometry optimization calculations. The potent ICT effect causes the derivatives to exhibit extended broad absorption with high absorption coefficients within the visible-NIR spectra region and concentration-dependent π–π-stacking-induced unusual fluorescence emission. Notably, this is a feasible way to extend the solar absorption range via the synchronous impact of the intrinsic π–π* transition of a large conjugated system and ICT transition. Moreover, the introduction of a quinoidal fragment on the bay region results in low-lying LUMO energy levels of C1 (−3.80 eV) and C2 (−3.83 eV), which indicates that the target perylene derivatives are efficient solar-harvesting compounds and potential candidate materials for use in photovoltaic devices.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grant no. 50573052 and 51173116) for supporting this research. We gratefully acknowledge Dr Xinchao Wang and Yulong Gong (Chongqing University) for the use of Gaussian software packages. We are also indebted to Mr Fansheng Cheng and Ms Qun Wang (Chengdu Green Energy and Green Manufacturing Technology R&D Center) for the opportunity to use the electrochemistry measurement workstation.Notes and references
- C.-Y. Chen, M.-K. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C.-h. Ngoc-le, J. D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103 CrossRef CAS PubMed; S.-J. Moon, E. Baranoff, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau, M. Grätzel and K. Sivula, Chem. Commun., 2011, 47, 8244 Search PubMed; X. Dang, J. Qi, M. T. Klug, P.-Y. Chen, D. S. Yun, N. X. Fang, P. T. Hammond and A. M. Belcher, Nano Lett., 2013, 13, 637 CrossRef PubMed.
- E. Ahmed, G. Ren, F. S. Kim, E. C. Hollenbeck and S. A. Jenekhe, Chem. Mater., 2011, 23, 4563 CrossRef CAS; Y. Zhou, L. Ding, K. Shi, Y.-Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957 CrossRef PubMed; R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215 CrossRef PubMed; T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025 CrossRef PubMed.
- Q. Yan, Z. Luo, K. Cai, Y. Ma and D. Zhao, Chem. Soc. Rev., 2014, 43, 4199 RSC.
- C. W. Struijk, A. B. Sieval, J. E. J. Dakhorst, M. van Dijk, P. Kimkes, R. B. M. Koehorst, H. Donker, T. J. Schaafsma, S. J. Picken, A. M. van de Craats, J. M. Warman, H. Zuilhof and E. J. R. Sudho1lter, J. Am. Chem. Soc., 2000, 122, 11057 CrossRef CAS.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939 CrossRef CAS PubMed; W. Liu, R. Tkachov, H. Komber, V. Senkovskyy, M. Schubert, Z. Wei, A. Facchetti, D. Neher and A. Kiriy, Polym. Chem., 2014, 1 Search PubMed.
- E. Yang, J. Wang, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2014, 118, 1630 CrossRef CAS PubMed; K. M. Lefler, C. H. Kim, Y.-L. Wu and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 1608 CrossRef.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435 CAS.
- L. Cao, Y.-Z. Wang, J.-Q. Zhong, Y.-Y. Han, W.-H. Zhang, X.-J. Yu, F.-Q. Xu, D.-C. Qi and A. T. S. Wee, J. Phys. Chem. C, 2014, 118, 4160 CAS.
- J. Li, F. Dierschke, J. Wu, A. C. Grimsdale and K. Müllen, J. Mater. Chem., 2006, 16, 96 RSC; M. Planells, F. J. Céspedes-Guirao, A. Forneli, Á. Sastre-Santos, F. Fernández-Lázaro and E. Palomares, J. Mater. Chem., 2008, 18, 5802 RSC; L. Zhao, T. Ma, H. Bai, G. Lu, C. Li and G. Shi, Langmuir, 2008, 24, 4380 CrossRef CAS PubMed.
- H. Langhals, A. Obermeier, Y. Floredo, A. Zanelli and L. Flamigni, Chem.–Eur. J., 2009, 15, 12733 CrossRef CAS PubMed.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Leec and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384 RSC.
- A. Keerthi, Y. Liu, Q. Wang and S. Valiyaveettil, Chem.–Eur. J., 2012, 18, 11669 CrossRef CAS PubMed; S. Mathew and H. Imahori, J. Mater. Chem., 2011, 21, 7166 RSC.
- F. Würthner, Chem. Commun., 2004, 1564 RSC; L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS PubMed.
- I. M. Blake, L. H. Rees, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 39, 1818 CrossRef CAS; W. Zeng, B. S. Lee, Y. M. Sung, K.-W. Huang, Y. Li, D. Kim and J. Wu, Chem. Commun., 2012, 48, 7684 RSC.
- S. Chai, S.-H. Wen and K.-L. Han, Org. Electron., 2011, 12, 1806 CrossRef CAS PubMed; J. Gao, C. Xiao, W. Jiang and Z. Wang, Org. Lett., 2014, 16, 394 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 03, revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- F. Würthner, V. Stepanenko, Z. Chen, C. R. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933 CrossRef PubMed.
- A. A. Vasilev, K. D. Mey, I. Asselberghs, K. Clays, B. Champagne, S. E. Angelova, M. I. Spassova, C. Li and K. Müllen, J. Phys. Chem. C, 2012, 116, 22711 CAS.
- C.-C. Chao and M.-k. Leung, J. Org. Chem., 2005, 70, 4323 CrossRef CAS PubMed.
- Á. J. Jiménez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608 RSC.
- S. Vajiravelu, L. Ramunas, G. J. Vidas, G. Valentas, J. Vygintasc and S. Valiyaveettil, J. Mater. Chem., 2009, 19, 4268 RSC.
- Y. Zhu, A. R. Rabindranath, T. Beyerlein and B. Tieke, Macromolecules, 2007, 40, 6981 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem.–Eur. J., 2001, 7, 2245 CrossRef.
- H. Langhals, S. Demmig and T. Potrawa, J. Prakt. Chem., 1991, 333, 733 CrossRef CAS PubMed; C. Burgdorff and H.-G. Löhmannsröben, Chem. Phys. Lett., 1992, 197, 358 CrossRef.
- A. D. Shaller, W. Wang, H. Gan and A. D. Q. Li, Angew. Chem., 2008, 120, 7819 CrossRef CAS PubMed; P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319 CrossRef PubMed.
- U. B. Cappel, M. H. Karlsson, N. G. Pschirer, F. Eickemeyer, J. Schöneboom, P. Erk, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2009, 113, 14595 CAS.
- Y. Li, Z. Pan, Y. Fu, Y. Chen, Z. Xie and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1663 CrossRef CAS PubMed.
- Y. Li, J. Zou, H.-L. Yip, C.-Z. Li, Y. Zhang, C.-C. Chueh, J. Intemann, Y. Xu, P.-W. Liang, Y. Chen and A. K.-Y. Jen, Macromolecules, 2013, 46, 5497 CrossRef CAS.
- S. Chen, Y. Liu, W. Qiu, X. Sun, Y. Ma and D. Zhu, Chem. Mater., 2005, 17, 2208 CrossRef CAS.
- H. Zhen, K. Li, Z. Huang, Z. Tang, R. Wu, G. Li, X. Liu and F. Zhang, Appl. Phys. Lett., 2012, 100, 213901 CrossRef PubMed.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864 CrossRef CAS.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246 CrossRef CAS PubMed.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS PubMed.
- Y. Zou, D. Gendron, R. Badrou-Aïch, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2009, 42, 2891 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum of Br-PDI, C1, C2 and C3. See DOI: 10.1039/c4ra07288j |
| This journal is © The Royal Society of Chemistry 2014 |