
Synthesis of N-urethane protected amino alkyl (S-methyl)-isothiouronium compounds and carbodiimide tethered peptidomimetics: an application for guanidino and substituted guanidino peptidomimetics synthesis†
Basavaprabhu Hosamania,
N. Narendrab,
Girish Prabhua and
Vommina V. Sureshbabu*a
aPeptide Research Laboratory, Department of Studies in Chemistry, Central College Campus, Bangalore University, #109, Dr. B. R. Ambedkar Veedhi, Bangalore 560 001, India. E-mail: sureshbabuvommina@rediffmail.com; hariccb@gmail.com; hariccb@hotmail.com; Tel: +91 80 22961339
bDepartment of Chemistry, University College of Science, Tumkur University, B. H. Road, Tumkur, India
First published on 18th September 2014
Abstract
The synthesis of N,N′-disubstituted and N,N′,N′′-trisubstituted guanidine linked peptidomimetic molecules suitably decorated in the peptide backbone has been delineated. Nα-Protected amino acid derived S-methyl isothiouronium derivatives are employed as the key intermediates for the synthesis of guanidinopeptide mimics. Synthesis of a new class of carbodiimide tethered dipeptidomimetics has also been outlined wherein a Staudinger-aza-Wittig type reaction between amino alkyl azide and isothiocyanato esters is employed. Thus obtained carbodiimides have been demonstrated as starting materials for the construction of guanidino peptide mimics as well as an array of trisubstituted guanidine mimetics bearing N-hydroxy, cyano and amino function as third substitutions at the guanidino unit in the backbone.
Introduction
Peptides are not ideal therapeutical agents per se. Their in vivo action is constrained by factors such as proteolytic degradation by proteases and poor cell membrane permeability. One prominent approach to improve the pharmaco-kinetic properties is to substitute peptides with synthetic analogs called peptide mimics1 and one such class is the backbone modified peptides.2 In these compounds the peptide backbone contains one or more unnatural linkages. Our group has described different classes of backbone modified peptides by inserting ureido,3 thioureido4 and selenoureido5 subunits as unnatural tethers. We now delineate our studies on the synthesis of guanidino peptide mimics under simple and mild conditions employing commonly used N-protectors namely Fmoc, Boc and Cbz groups.A major drawback of most of the non-native linkages is that they tend to decrease the solubility of the pseudo-peptides in aqueous solvents as well as cause aggregation in such solvents. The problem has been partly addressed by the incorporation into the peptide backbone of guanidino group/s, which remains protonated at physiological pH and hence renders hydrophilicity.6
This is due to the strong basicity of guanidine moiety which ensures protonation over a wide pH range. Also guanidinium functionality is useful as strong and selective anion-binding site in receptor designs for small oxoanionic target molecules or π-electron rich aromatic moieties of substrates.7
The oligomeric guanidines may be valuable in the design of protein ligands wherein the guanidine backbone may interact with target proteins by electrostatic interactions and/or hydrogen bonds.8 Also the replacement of one or more peptide bonds with unnatural linkages tend to improve the pharmacokinetic properties of the bioactive peptides.9
Though several amino acid derived guanidines (Fig. 1) exist in the literature, the insertion of guanidine into a peptide backbone are limited to two reports, wherein Pbf-activated thiourea and di-Boc-isothiouronium derivatives were utilized for guanidinylation. Fan et al.,8 reported the solid phase as well as solution phase synthesis of oligomeric guanidines by making use of Pbf-activated thiourea as monomer units (Fig. 2).
 | ||
| Fig. 1 Examples of amino acid derived guanidines. | ||
 | ||
| Fig. 2 Reported oligomeric guanidines synthesized in N → C direction (B) and C → N direction (C). | ||
The other one delineates the synthesis of dipeptide consisting of α- and γ-amino acid with guanidinium group as an amide bond replacement in the main chain employing 1,3-di-Boc-2-methyl-isothiourea as guanidinylating agent.7
Several methods have been developed for the preparation of substituted guanidines in solution.10,11 The reaction of amines with S-alkylisothioureas (Rathke procedure) is an alternative to the use of guanidinylating agents (Fig. 3) for the synthesis of substituted guanidines.12 Here, the reaction involves nucleophilic displacement of the alkyl thiol via an addition–elimination pathway. The S-alkylisothiouronium salt can be easily prepared by treatment of thiourea with an alkylating agent.
 | ||
| Fig. 3 Thiourea and non-thiourea based guanylating reagents. | ||
In the present study, we describe the preparation of N-protected amino acid derived isothiouronium derivatives as guanylating agents and their utility in the preparation of guanidinopeptides under mild conditions. Isothiouronium compounds can serve as important intermediates for the synthesis of thiols.13a Also they are reported to be useful in the condensation reactions with aldehyde and amine to yield triazine derivatives.13b
Results and discussion
N-Protected amino acid derived S-methyl isothiouronium derivatives [Pg-Xaa-ψCH2-N-C(SMe)-NH2] 3
In the initial part of the study, the feasibility of thioureidopeptides to serve as the precursors for guanidine tethered peptide mimics was explored. The reaction of thioureidopeptides with ammonia or NH4Cl in presence of HgCl2 and Mukaiyama reagent was not successful. Consequently, the other route comprising of the S-methyl isothiouronium derivative of amino acids was conceived. Isothiouronium compounds are a class of amino acid derivatives that can be made starting from Nα-protected amino alkyl isothiocyanates and subsequently utilized as key intermediates for the desired target molecules.Monosubstituted thiourea derivatives can be accessed through the reaction of primary amines with thiocyanates such as KSCN, NH4SCN14 or coupling with benzoyl isothiocyanate followed by deprotection of benzoyl group.15 Stadlwieser et al.,16 reported the synthesis of resin bound amino ester derived thioureas, wherein amino acid ester was treated with allyloxycarbonyl (Alloc)-isothiocyanate followed by the removal of Alloc group.
Initially, Nα-protected amino alkyl monosubstituted thioureas 2 were obtained by the reaction of Nα-protected amino alkyl isothiocyanate4 with NH4HCO3 (ref. 17) in THF/H2O at 0 °C (Scheme 1). The reaction is clean and complete conversion of isothiocyanate to thiourea was observed within 2–3 h and crude products were then purified through column chromatography (compounds 2a–h; Table 1). The products were characterized by 1H and 13C NMR analyses. The chiral HPLC profile of Fmoc-L-Phg-ψ[CH2-NH-CS-NH2] 2e and Fmoc-D-Phg-ψ[CH2-NH-CS-NH2] 2e′ made from L-Phg and D-Phg (the amino acid with high tendency to lose optical integrity) showed only one peak Rt = 25.85 min and 29.81 min respectively, (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 n-hexane/isopropanol in isocratic mode in 40 min). Co-injection of 2e and 2e′ showed two well separated peaks (Rt = 25.31 and Rt = 30.21 min). Thus it can be concluded that the compounds 2 made via the present protocol were optically pure. Also, the synthesized compounds were found to be shelf stable to storage for several weeks at room temperature.
:20 n-hexane/isopropanol in isocratic mode in 40 min). Co-injection of 2e and 2e′ showed two well separated peaks (Rt = 25.31 and Rt = 30.21 min). Thus it can be concluded that the compounds 2 made via the present protocol were optically pure. Also, the synthesized compounds were found to be shelf stable to storage for several weeks at room temperature.
 | ||
| Scheme 1 | ||
| Entry | Thiourea | [α]25D (c 1, CHCl3) | Yield (%) | Entry | Isothiouronium derivative | [α]25D (c 1, CHCl3) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 2a | Fmoc-Val-ψCH2-NH-C(S)-NH2 | 30.16 | 81 | 3a | Fmoc-Val-ψCH2-N-C(SMe)-NH2 | 42.3 | 88 |
| 2b | Fmoc-Leu-ψCH2-NH-C(S)-NH2 | 26.08 | 84 | 3b | Fmoc-Leu-ψCH2-N-C(SMe)-NH2 | 62.4 | 90 |
| 2c | Fmoc-Gly-ψCH2-NH-C(S)-NH2 | — | 82 | 3c | Fmoc-Gly-ψCH2-N-C(SMe)-NH2 | — | 89 |
| 2d | Fmoc-Pro-ψCH2-NH-C(S)-NH2 | 41.04 | 79 | 3d | Fmoc-Pro-ψCH2-N-C(SMe)-NH2 | 16.3 | 86 |
| 2e | Fmoc-Phg-ψCH2-NH-C(S)-NH2 | −29.2 | 72 | 3e | Fmoc-Phg-ψCH2-N-C(SMe)-NH2 | −21.3 | 82 |
| 2f | Cbz-Ile-ψCH2-NH-C(S)-NH2 | 21.4 | 73 | 3f | Cbz-Ile-ψCH2-N-C(SMe)-NH2 | 4.6 | 88 |
| 2g | Boc-Phe-ψCH2-NH-C(S)-NH2 | −5.04 | 88 | 3g | Boc-Phe-ψCH2-N-C(SMe)-NH2 | −1.1 | 96 |
| 2h | Boc-Ala-ψCH2-NH-C(S)-NH2 | 38.12 | 94 | 3h | Boc-Ala-ψCH2-N-C(SMe)-NH2 | 18.5 | 98 |
Compounds 2 and iodomethane were then refluxed in acetone to obtain corresponding S-methyl isothiouronium derivative 3 as the salts of HI (compounds 3a–h; Scheme 1 and Table 1). The reaction was complete within an hour affording the products in excellent yield. Evaporation of the solvent was sufficient to isolate the pure products.
N-protected amino acid derived guanidinopeptide mimics [Pg-Xaa-ψCH2-NH-C(NH)-NH-Xbb-COOY] 4a–c, 4e, 4g and h
The S-methylisothiouronium salts 3 were deprotonated with 1.0 equiv of NMM and then treated with amino acid ester in acetonitrile. The reaction mixture was heated at 60 °C to obtain the crude (Scheme 1). Thus obtained products were column chromatographed using neutral alumina to obtain pure products (compounds 4a–c, 4e, 4g and h; Table 2). The compounds were then characterized by mass spectrometry, 1H and 13C NMR analyses.| Entry | Pg | R1 | R2 | X | Yield (%) |
|---|---|---|---|---|---|
| 4a | Fmoc | –CH(CH3)2 | –CH3 | Me | 59 |
| 4b | Fmoc | –CH2CH(CH3)2 | –CH2CH2 | Me | 66 |
| 4c | Fmoc | –H | –CH2C6H5 | Me | 63 |
| 4e | Fmoc | –C6H5 | –CH3 | Me | 61 |
| 4g | Boc | –CH2C6H5 | –CH(CH3)2 | Me | 71 |
| 4h | Boc | –CH3 | –CH(CH3)2 | Et | 68 |
Test for epimerisation
Fan et al., have not emphasized on establishing the optical purity of the monomers as well as the oligomeric guanidines. The optical purity of the N-protected guanidinopeptide mimic was evaluated by the 1H NMR study of the model compounds 4e and 4e′ prepared via the present protocol is illustrated in Fig. 4. The diastereomeric guanidinopeptide mimics 4e and 4e′ were synthesized by coupling Fmoc-Phg-ψ[CH2NC(SMe)NH2] 3e with L- and D-Ala-OMe respectively in separate experiments. 1H NMR of 4e and 4e′ showed a single distinct methyl group doublet. Observed δ values for the –CH3 group of the compounds are, 4e: 1.17, 1.18 and 4e′: 1.24, 1.25 ppm. Further, the mixture prepared by coupling 3e with racemic L,D-Ala-OMe showed methyl groups as two doublets with δ values 1.15, 1.16 and 1.23, 1.24 indicating the presence of two isomers and confirming that the protocol is free from epimerisation. The optical purity of compounds 4e and 4e′ was demonstrated to be high through chiral HPLC studies.18 | ||
| Fig. 4 Diastereomeric guanidinodipeptide mimics 4e and 4e′ synthesized for epimerisation studies. | ||
Synthesis of N-protected carbodiimide tethered dipeptidomimetics [Pg-Xaa-ψCH2-(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CN)-Xbb-COOY] 5
CN)-Xbb-COOY] 5
For the synthesis of N,N′,N′′-trisubstituted guanidino peptide mimics such as, N-hydroxyguanidines,19 aminoguanidines20 and cyanoguanidines,21 an alternative route need to be envisaged. Thus, we extended our study to develop a common protocol for the synthesis of diverse classes of trisubstituted guanidines. To access these molecules one of the best routes is the reaction of a suitable substituted amine with a carbodiimide.22
The envisaged backbone carbodiimide mimics are itself a novel class of peptide mimetics. Previously peptidic carbodiimides were obtained in situ by the coupling of peptidyl amine with aryl isothiocyanate followed by desulfurization.23,24 Carbodiimide linked amino esters illustrated in Fig. 5 were prepared through the reaction of azido ester with aryl isocyanates or by the dehydration of corresponding urea, which were N-terminal derived ones.25–27
 | ||
| Fig. 5 Reported amino acid derived carbodiimides and peptidic carbodiimides. | ||
Fan et al.,8 carried out the oligomeric guanidine synthesis employing Pbf-activated thiourea as monomer units and performed the guanidinylation in presence of EDC, wherein carbodiimides were key intermediates.
As we aimed to introduce carbodiimide at C-terminus of peptide backbone, we sought to employ Staudinger-aza-Wittig type reaction using N-protected amino alkyl azides and isothiocyanato esters. The intermolecular aza-Wittig type reaction of iminophosphoranes with heterocumulenes such as isocyanates/isothiocyanates can be carried out under neutral conditions28 and thus amenable to the preparation of carbodiimide tethered peptidomimetics. Isothiocyanato esters undergo desired transformation with the extrusion of triphenylphosphinethioxide leading to hitherto unreported class of N-protected carbodiimide tethered dipeptidomimetics, which can be satisfactorily converted into guanidinopeptide mimics with diverse substitutions as demonstrated below.
In a typical experiment, Cbz-Phe-CH2OH derived amino alkyl azide4 was treated with triphenylphosphine in dry CH2Cl2 at 0 °C and then allowed to reach room temperature. Upon disappearance of the azide (TLC), alaninyl isothiocyanato ester29 was added and the mixture was stirred for 2–3 h (Scheme 2). The IR analysis of the crude reaction mixture showed a strong absorption band at 2118 cm−1 corresponding to –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN– stretch of the carbodiimide30 (see characterisation data for compounds 5a–f). Carbodiimides could not be purified through silica gel column chromatography due to their conversion to the corresponding ureas (∼75%). However careful purification through neutral alumina afforded the desired carbodiimide 5d. Employing the same protocol, a series of carbodiimides, 5a–c as illustrated in Table 3 were then prepared, isolated and characterized.
CN– stretch of the carbodiimide30 (see characterisation data for compounds 5a–f). Carbodiimides could not be purified through silica gel column chromatography due to their conversion to the corresponding ureas (∼75%). However careful purification through neutral alumina afforded the desired carbodiimide 5d. Employing the same protocol, a series of carbodiimides, 5a–c as illustrated in Table 3 were then prepared, isolated and characterized.
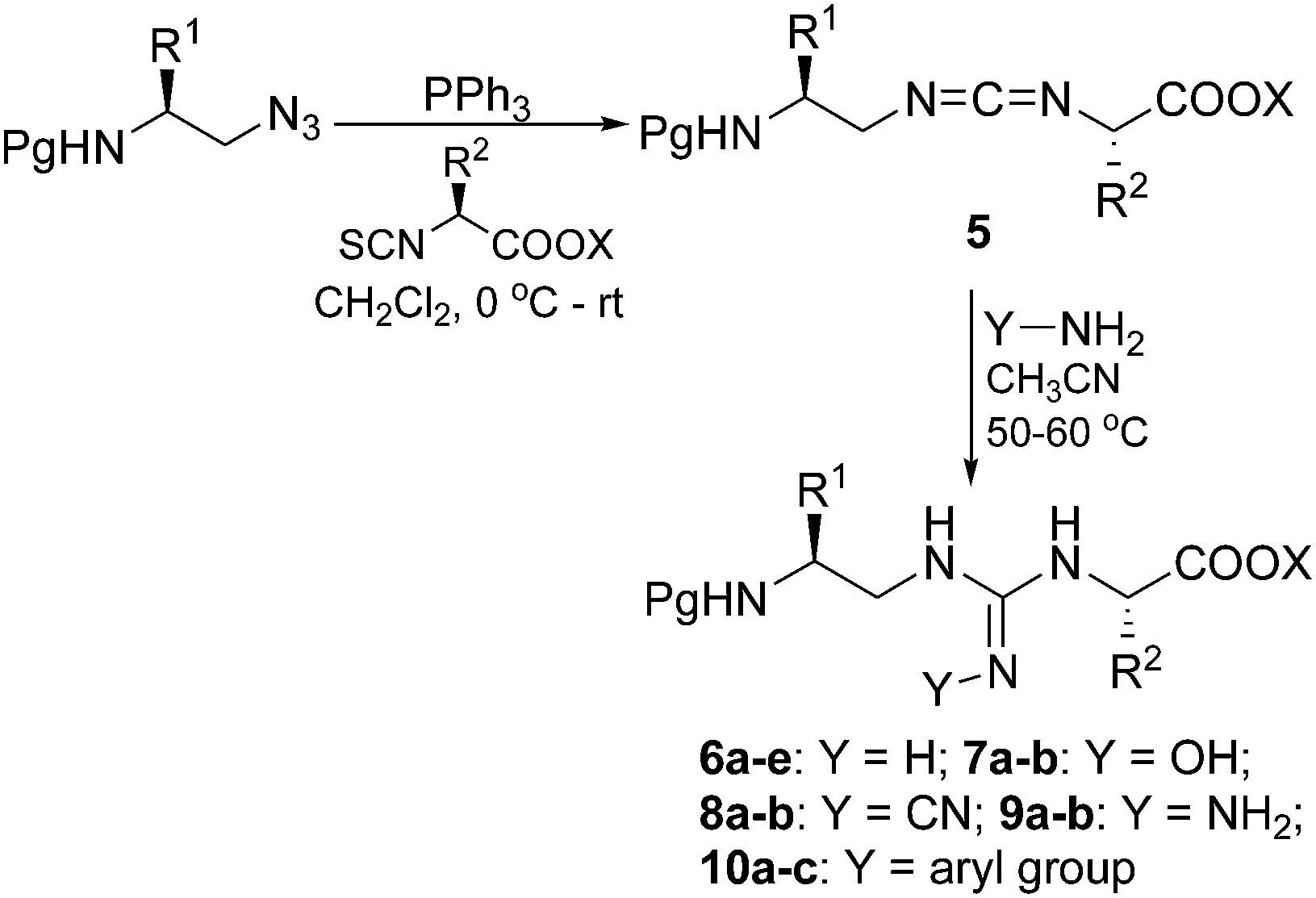 | ||
| Scheme 2 | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Pg | R1 | R2 | X | Yield (%) |
| 5a | Fmoc | –CH3 | –CH(CH3)2 | Me | 71 |
| 5b | Cbz | –CH2COOBzl | –H | Bzl | 70 |
| 5c | Cbz | –(CH2)2COOBzl | –CH2C6H5 | Et | 68 |
| 5d | Cbz | –CH2C6H5 | –CH3 | Me | 75 |

Carbodiimides were confirmed to be optically pure by carrying out 1H NMR analysis of the model compounds 5e and 5f. These were prepared starting from Cbz-Phe-ψ[CH2-N3] with isothiocyanates derived from (R)- and (S)-1-phenethylamine respectively. 1H NMR of the crude 5e and 5f showed a single distinct methyl group doublet. Observed δ values for the –CH3 group are as follows: 5e: 1.48, 1.49 and 5f: 1.54, 1.55. The equimolar mixture of 5e and 5f showed two distinct doublets at δ 1.46, 1.48, 1.52, 1.54, suggesting the presence of two isomers (see pages S47–48 of ESI†). This study infers the absence of epimerisation during their synthesis from corresponding azides.
N,N′-Disubstituted and N,N′,N′′-trisubstituted guanidine tethered dipeptidomimetics [Pg-Xaa-ψCH2-NH-C(NY)-NH-Xbb-COOX] 6a–e, 7–10
Carbodiimide prepared as in the above protocol was treated with NH4Cl in acetonitrile at 50–60 °C to afford the guanidinopeptides 6a–e (Scheme 2 and Table 4). Further, the synthetic utility of the carbodiimide as useful intermediate was also demonstrated by its reaction with hydroxylamine to yield hydroxyguanidines (compounds 7a and b, Table 4), sodium cyanamide to yield cyanoguanidine (compounds 8a and b, Table 4), hydrazine to yield aminoguanidine (compounds 9a and b, Table 4), and aryl amines to yield N,N′,N′′-trisubstituted guanidine (compounds 10a–c, Table 4) as illustrated in Scheme 2. Acetonitrile serve as a good solvent among the solvents screened such as DMF, THF and 1,4-dioxane and the reaction requires higher temperature to provide desired products in moderate to good yields. The products were purified on neutral alumina column chromatography and the compounds were confirmed by mass spectrometry, 1H and 13C NMR analyses.| Entry | Pg | R1 | R2 | Y | X | Yield (%) |
|---|---|---|---|---|---|---|
| 6a | Cbz | –CH2C6H5 | –CH3 | H | Me | 69 |
| 6b | Cbz | –CH(CH3)2 | –CH2CH(CH3)2 | H | Me | 67 |
| 6c | Cbz | –CH2S(Bzl) | –CH3 | H | Me | 59 |
| 6d | Fmoc | –CH3 | –CH2CH2 | H | Me | 63 |
| 6e | Fmoc | –CH2C6H5 | –CH2CH2 | H | Me | 58 |
| 7a | Fmoc | –CH3 | –CH(CH3)2 | OH | Me | 56 |
| 7b | Fmoc | –CH2C6H5 | –CH2C6H5 | OH | Me | 53 |
| 8a | Fmoc | –CH2C6H5 | –H | CN | Et | 64 |
| 8b | Fmoc | –CH2C6H5 | –CH2CH(CH3)2 | CN | Bzl | 67 |
| 9a | Cbz | –CH2CH(CH3)2 | –CH2C6H5 | NH2 | Me | 71 |
| 9b | Fmoc | –CH3 | –CH(CH3)2 | NH2 | Me | 51 |
| 10a | Fmoc | –CH3 | –CH(CH3)CH2CH3 | C6H5 | Me | 65 |
| 10b | Fmoc | –H | –CH2CH(CH3)2 | CH2C6H5 | Me | 58 |
| 10c | Fmoc | –CH3 | –CH(CH3)2 | 4-Cl–C6H4 | Me | 62 |
Conclusion
We have demonstrated the synthesis of Nα-protected amino alkyl S-methyl isothiouronium derivatives using a variation of Rathke protocol and accomplished the synthesis of novel class of carbodiimide tethered dipeptidomimetics through a Staudinger-aza-wittig type reaction. These compounds were then demonstrated as intermediates for the synthesis of guanidine tethered backbone modified peptidomimetics. A range of guanidinium derivatives such as free guanidine, N-hydroxy, cyano, amino and N,N′,N′′-trisubstituted guanidinium units were accomplished under mild experimental conditions so as to obtain them in good yields. The access to guanidinium and its derivatives serves to expand the repertoire of these unique entities in drug and medicinal fields as they mimic the biological roles played by Arg and its variants.Experimental section
General procedure for the synthesis of Nα-protected amino alkyl thioureas 2a–h
To a stirred solution of Nα-protected amino alkyl isothiocyanate (1.0 mmol) in THF (5.0 mL) was added 3.0 eq of ammonium bicarbonate in 3.0 mL of water. The reaction mixture was cooled to 0 °C and stirred further for 3 h. After completion of the reaction as monitored through TLC, the reaction mixture was concentrated and diluted with EtOAc. The organic layer was washed with water (10 mL) and brine (5 mL) and dried over anhydrous Na2SO4. The organic layer was evaporated in vacuo to afford the product 2 in quantitative yield.General procedure for the synthesis of Nα-protected amino alkyl isothiouronium derivatives 3a–h
To a solution of Nα-protected amino alkyl thiourea 2 (1.0 mmol) in acetone (8.0 mL) was added methyl iodide (1.5 mmol) and the reaction mixture was heated at reflux. The reaction was completed in 1–2 h as judged by TLC. Then the solvent was evaporated in vacuo and the resultant product 3 was obtained in excellent yield.General procedure for the synthesis of guanidine tethered peptidomimetics 4a–c, 4e, 4g and h
To a neutralized solution of isothiouronium derivative 3 (1.0 mmol) in acetonitrile (6.0 mL) was added amino acid ester (1.3 mmol). The resultant mixture was heated at 60 °C till the completion of the reaction as monitored through TLC. After completion of the reaction, solvent was evaporated in vacuo and the obtained crude product 4 was then purified through column chromatography. [Caution: reaction has to be carried out in a fume hood as it involves the expulsion of toxic methyl mercaptan (CH3SH) as by-product].General procedure for the synthesis of carbodiimide 5a–f
To a solution of N-protected amino alkyl azide in dry CH2Cl2 at 0 °C was added triphenylphosphine (1.2 mmol). The reaction mixture was stirred for 10–15 min. then requisite isothiocyanate (1.3 mmol) was added and the mixture was stirred for about 2–3 h at same temperature till the completion of the reaction. The progress of the reaction was monitored through IR, where it showed a strong absorption band in the region 2116–2130 cm−1 characteristic of carbodiimide 5. The solvent was evaporated under reduced pressure and the product was purified through neutral alumna to afford the desired carbodiimide 5.General procedure for the synthesis of guanidine tethered peptidomimetics, 6a–e
To a solution of N-protected amino alkyl azide in dry CH2Cl2 at 0 °C was added triphenylphosphine (1.2 mmol). The reaction mixture was stirred for 10–15 min. then isothiocyanato ester (1.3 mmol) was added and the mixture was stirred for about 2–3 h at same temperature till the completion of the reaction. The progress of the reaction was monitored through IR, where it showed a strong absorption band in the region 2116–2130 cm−1 characteristic of carbodiimide. The solvent was evaporated under reduced pressure and then the product without isolation was treated with NH4Cl (2.0 mmol) in acetonitrile. The reaction mixture was heated at 60 °C till the completion of the reaction as monitored through TLC. The organic layer was then evaporated in vacuo to afford the crude product 6 in quantitative yield. The product was then purified through column chromatography.General procedure for the synthesis of N-hydroxyguanidine (7a and b), aminoguanidine (8a and b) and cyanoguanidines (9a and b) and trisubstituted guanidine mimetics (10a–c)
To a solution of N-protected amino alkyl azide in dry CH2Cl2 at 0 °C was added triphenylphosphine (1.2 mmol). The reaction mixture was stirred for 10–15 min. then isothiocyanato ester (1.3 mmol) was added and the mixture was stirred for about 2–3 h at same temperature till the completion of the reaction. The progress of the reaction was monitored through IR, where it showed a strong absorption band in the region 2116–2130 cm−1 characteristic of carbodiimide. The solvent was evaporated under reduced pressure and then the product without isolation was treated with a 1.5 mmol solution of hydroxylamine in acetonitrile to yield hydroxyguanidines (compounds 7a and b), sodium cyanamide in acetonitrile to yield cyanoguanidine (compounds 8a and b), hydrazine in acetonitrile to yield aminoguanidines (compounds 9a and b), and aryl amines in acetonitrile to yield N,N′,N′′-trisubstituted guanidines (compounds 10a–c). The resulting mixture was heated at 60 °C till the completion of the reaction as monitored by TLC. Then the organic layer was evaporated in vacuo to afford the crude products in quantitative yield. The products were then purified through column chromatography.(9H-Fluoren-9-yl)methyl-3-methyl-1-thioureidobutan-2-ylcarbamate, Fmoc-Val-ψCH2-NH-C(S)-NH2 (2a)
Yield 81%; 1H NMR (400 MHz, DMSO-d6) δ 0.89 (d, J = 6.7 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 2.11 (m, 1H), 3.45 (m, 2H), 4.14–4.23 (m, 2H), 4.44 (m, 2H), 5.34 (br, s, 1H), 6.14 (s, br, 1H), 7.33 (t, J = 7.2 Hz, 2H), 7.41 (t, J = 7.2 Hz, 2H), 7.61 (m, 2H), 7.78 (d, J = 7.2 Hz, 2H), 8.02 (s, br, 2H); 13C NMR (100 MHz, DMSO-d6) δ 18.05, 19.16, 30.39, 45.23,47.01, 55.84, 65.27, 120.12, 120.15, 127.09, 127.12, 127.61, 127.64, 140.72, 140.75, 143.81, 143.97, 156.24, 183.40; HRMS calcd for C21H25N3O2S m/z 406.1554 (M+ + Na), found 406.1556.(9H-Fluoren-9-yl)methyl-4-methyl-1-thioureidobutan-2-ylcarbamate, Fmoc-Leu-ψCH2-NH-C(S)-NH2 (2b)
Yield 84%; 1H NMR (400 MHz, DMSO-d6) δ 0.91 (d, J = 6.2 Hz, 6H), 1.32–1.78 (m, 3H), 3.38 (d, J = 6.8 Hz, 2H), 4.12–4.28 (m, 2H), 4.43 (m, 2H), 5.31 (br s, 1H), 6.10 (s, br, 1H), 7.28 (t, J = 7.4 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.57 (t, J = 7.4 Hz, 2H), 7.73 (d, J = 7.4 Hz, 2H), 8.01 (br s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 23.22, 23.31, 24.21, 41.42, 46.80, 48.13, 49.02, 65.10, 120.10, 120.13, 125.20, 127.04, 127.07, 127.59, 140.73, 140.75, 143.77, 143.99, 155.87, 183.44; HRMS calcd for C22H27N3O2S m/z 420.1836 (M+ + Na), found 420.1844.(9H-Fluoren-9-yl)methyl-2-thioureidoethylcarbamate, Fmoc-Gly-ψCH2-NH-C(S)-NH2 (2c)
Yield 82%; 1H NMR (400 MHz, DMSO-d6) δ 3.16 (m, 2H), 3.65 (m, 2H), 4.22 (t, J = 6.6 Hz, 1H), 4.45 (d, J = 6.6 Hz, 2H), 5.27 (s, br, 1H), 6.12 (s, br, 1H), 7.28–7.43 (m, 4H), 7.60 (m, 2H), 7.77 (d, J = 7.5 Hz, 2H), 8.05 (s, br, 2H); 13C NMR (100 MHz, DMSO-d6) δ 40.11, 43.68, 46.90, 65.41, 120.14, 125.18, 127.10, 127.63, 140.73, 143.88, 156.24, 183.40; HRMS calcd for C18H19N3O2S m/z 364.4170 (M+ + Na), found 364.4177.(9H-fluoren-9-yl)methyl-2-(thioureidomethyl)pyrrolidine-1-carboxylate, Fmoc-Pro-ψCH2-NH-C(S)-NH2 (2d)
Yield 79%; 1H NMR (400 MHz, DMSO-d6) δ 1.72–2.02 (br, 4H), 3.18–3.54 (m, 4H), 3.99–4.44 (m, 4H), 6.18 (s, br, 1H), 7.26–7.75 (m, 8H), 8.11 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 23.00, 28.32, 45.68, 46.90, 56.27, 57.30, 66.32, 120.13, 125.13, 125.28, 127.15, 127.67, 140.66, 140.77, 143.78, 154.20, 183.69; HRMS calcd for C21H23N3O2S m/z 404.1412 (M+ + Na), found 404.4804.(9H-Fluoren-9-yl)methyl-1-phenyl-2-thioureidoethyl-carbamate, Fmoc-Phg-ψCH2-NH-C(S)-NH2 (2e)
Yield 72%; 1H NMR (400 MHz, DMSO-d6) δ 3.21 (m, 2H), 4.01 (m, 1H), 4.15 (t, J = 6.8 Hz, 1H), 4.37 (d, J = 8.0 Hz, 2H), 5.52 (s, br, 1H), 6.17 (s, br, 1H), 7.20–7.80 (m, 13H), 8.19 (s, br, 2H); 13C NMR (100 MHz, DMSO-d6) δ 47.67, 50.04, 55.40, 67.54, 120.55, 125.53, 127.02, 127.64, 128.27, 129.18, 129.67, 134.00, 137.96, 141.83, 144.15, 155.76, 182.86; HRMS calcd for C24H23N3O2S m/z 440.1435 (M+ + Na), found 404.1439.Benzyl-3-methyl-1-thioureidopentan-2-ylcarbamate, Cbz-Ile-ψCH2-NH-C(S)-NH2 (2f)
Yield 73%; 1H NMR (400 MHz, CDCl3) δ 0.97 (m, 6H), 1.18 (m, 1H), 1.49 (m, 1H), 1.80 (m, 1H), 3.15 (m, 2H), 4.14 (m, 1H), 5.38 (s, br, 1H), 6.08 (s, br, 1H), 7.30–7.49 (m, 5H), 7.89 (s, br, 2H); 13C NMR (100 MHz, CDCl3) δ 11.35, 15.34, 25.21, 37.58, 47.17, 56.13, 66.84, 127.55, 128.08, 128.55, 136.26, 157.56, 183.95; HRMS calcd for C15H23N3O2S m/z 332.1390 (M+ + Na), found 332.1392.Tert-Butyl-3-phenyl-1-thioureidopropan-2-ylcarbamate, Boc-Phe-ψCH2-NH-C(S)-NH2 (2g)
Yield 88%; 1H NMR (400 MHz, CDCl3) δ 1.49 (s, 9H), 3.05 (2 dd, J = 13.7 and 6.9 Hz, 2H), 3.18 (m, 2H), 4.43 (m, 1H), 5.18 (br, s, 1H), 6.14 (s, br, 1H), 7.21–7.36 (m, 5H), 7.98 (s, br, 2H); 13C NMR (100 MHz, CDCl3) δ 28.22, 39.28, 51.03, 53.40, 80.04, 128.47, 128.75, 128.83, 129.08, 156.54, 183.92; HRMS calcd for C15H23N3O2S m/z 332.1246 (M+ + Na), found 332.1250.Tert-Butyl-1-thioureidopropan-2-ylcarbamate, Boc-Ala-ψCH2-NH-C(S)-NH2 (2h)
Yield 94%; 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 9H), 1.58 (d, J = 7.1 Hz, 3H), 3.01 (m, 2H), 4.23 (m, 1H), 5.31 (s, br, 1H), 6.07 (s, br, 1H), 7.82 (s, br, 2H); 13C NMR (100 MHz, CDCl3) δ 18.41, 30.18, 41.17, 50.54, 80.93, 155.53, 181.19; HRMS calcd for C9H19N3O2S m/z 256.1076 (M+ + Na), found 256.1080.(S,Z)-(9H-Fluoren-9-yl)methyl-1-(amino(methylthio)methyl-eneamino)3-methylbutan-2-ylcarbamate, Fmoc-Val-ψCH2-NH-C(SMe)-NH2 (3a)
Yield 88%; 1H NMR (400 MHz, CDCl3) δ 0.98 (d, J = 7.81 Hz, 6H), 1.91 (m, 1H), 2.23 (s, 2H), 3.08 (m, 2H), 3.52 (m, 1H), 4.20 (t, J = 13.28 Hz, 1H), 4.44 (d, J = 6.72 Hz, 2H), 5.25 (s, br, 1H), 6.13 (s, br, 1H), 7.30 (t, J = 14.78 Hz, 2H), 7.40 (t, J = 14.7 Hz, 2H), 7.58 (d, J = 7.32 Hz, 2H), 7.76 (d, J = 7.44 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 19.64, 29.64, 41.08, 47.06, 56.05, 67.03, 119.86, 119.97, 125.33, 127.10, 127.63, 141.15, 143.54, 143.70, 143.89, 14.78, 156.36, 168.77; HRMS calcd for C22H27N3O2S m/z 398.1904 (M+ + H), found 398.1906.(S,Z)-(9H-Fluoren-9-yl)methyl-1-(amino(methylthio)methyl-eneeamino)4-methylpentan-2-ylcarbamate, Fmoc-Leu-ψCH2-NH-C(SMe)-NH2 (3b)
Yield 90%; 1H NMR (400 MHz, CDCl3) δ 0.92 (dd, J = 2.28 Hz, J = 6.2 Hz, 6H), 1.32 (m,1H), 1.63 (m, 2H), 2.19 (s, 3H), 3.51 (m, 1H), 3.68 (m, 1H), 3.76 (m, 1H), 4.21 (t, J = 6.88 Hz, 1H), 4.45 (d, J = 6.88 Hz, 2H), 5.24 (s, br, 1H), 6.19 (s, br, 1H), 7.32 (t, J = 7.32 Hz, 2H), 7.40 (t, J = 7.32 Hz, 2H), 7.59 (d, J = 7.32 Hz, 2H), 7.77 (d, J = 7.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.64, 22.96, 24.72, 40.44, 47.03, 48.47, 51.60, 66.93, 119.85, 119.96, 125.36, 127.10, 127.63, 127.82, 141.15, 143.35, 143.69, 143.84, 156.06, 168.78; HRMS calcd for C30H29N3O2S m/z 412.2070 (M+ + H), found 412.2071.(Z)-(9H-Fluoren-9-yl)methyl-2-(amino(methylthio)methylene-amino)ethylcarbamate, Fmoc-Gly-ψCH2-NH-C(SMe)-NH2 (3c)
Yield 89%; 1H NMR (400 MHz, CDCl3) δ 2.10 (s, 3H), 3.42 (m, 2H), 3.52 (m, 2H), 4.19 (t, J = 12.66 Hz, 1H), 4.42 (d, J = 6.57 Hz, 2H), 5.28 (s, br, 1H), 6.12 (s, br, 1H), 7.29 (t, J = 14.45 Hz, 2H), 7.38 (t, J = 14.59 Hz, 2H), 7.57 (d, J = 7.08 Hz, 2H), 7.75 (d, J = 7.38 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.92, 38.76, 44.37, 46.86, 67.46, 119.89, 125.12, 125.25, 127.09, 127.65, 127.73, 141.02, 143.41, 143.62, 158.32, 168.85; HRMS calcd for C19H21N3O2S m/z 356.1442 (M+ + H), found 356.1440.(S,Z)-(9H-Fluoren-9-yl)methyl-2-(amino(methylthio)methyl-eneamino)methyl)pyrrolidines-1-carboxylate, Fmoc-Pro-ψCH2-NH-C(SMe)-NH2 (3d)
Yield 82%; 1H NMR (400 MHz, CDCl3) δ 1.29 (m, 4H), 2.10 (s, 3H), 3.23 (m, 2H), 3.52 (m, 1H), 3.66 (m, 2H), 4.23 (t, J = 7.0 Hz, 1H), 4.44 (m, 2H), 6.10 (s, br, 1H), 7.31–7.43 (m, 4H), 7.60 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.07, 23.66, 29.36, 47.19, 50.77, 54.89, 56.18, 68.10, 119.93, 120.06, 124.87, 127.07, 127.68, 127.89, 141.25, 143.26, 143.53, 143.79, 157.82, 168.56; HRMS calcd for C21H26N3O2S m/z 396.1750 (M+ + H), found 396.1748.(S,Z)-(9H-Fluoren-9-yl)methyl-2-(amino(methylthio)methyleneamino)1-phenylethyl-carbamate, Fmoc-Phg-ψCH2-NH-C(SMe)-NH2 (3e)
Yield 88%; 1H NMR (400 MHz, CDCl3) δ 2.14 (s, 3H), 3.47 (m, 2H), 4.10–4.40 (m, 4H), 5.16 (s, br, 1H), 6.18 (s, br, 1H), 7.20–7.78 (m, 13H); 13C NMR (100 MHz, CDCl3) δ 14.75, 39.05, 47.15, 55.23, 66.92, 124.86, 126.45, 127.06, 127.70, 128.47, 128.74, 129.17, 138.12, 141.28, 143.62, 158.06, 168.28; HRMS calcd for C25H25N3O2S m/z 432.2030 (M+ + H), found 432.2031.Benzyl(2S,3R)-1-(Z-amino(methylthio)methyleneamino)3-methylpentan-2-ylcarbamate, Cbz-Ile-ψCH2-NH-C(SMe)-NH2 (3f)
Yield 96%; 1H NMR (400 MHz, CDCl3): δ 0.75–1.26 (m, 9H), 3.13–3.39 (m, 2H), 4.10 (br, 1H), 4.30 (br, 2H), 4.63 (br, 1H), 6.20 (br, 1H), 7.31–7.77 (m, 8H), 8.14 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 12.93, 14.15, 15.40, 25.21, 41.37, 49.72, 49.83, 67.54, 128.77, 128.95, 129.08, 132.80, 133.78, 136.58, 157.94, 168.14; HRMS calcd for C16H25N3O2S m/z 324.1736 (M+ + H), found 324.1738.(S,Z)-tert-Butyl-1-(amino(methylthio)methyleneamino)3-phenylpropan-2-ylcarbamate, Boc-Phe-ψCH2-NH-C(SMe)-NH2 (3g)
Yield 98%; 1H NMR (400 MHz, CDCl3) δ 1.42 (s, 9H), 2.96 (m, 2H), 3.67 (d, J = 3.6 Hz, 2H), 4.02 (m, 1H), 5.10 (s, br, 1H), 6.19 (s, br, 1H), 7.12–7.37 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 15.13, 28.32, 38.04, 46.76, 51.22, 80.10, 128.71, 128.77, 129.18, 129.37, 136.65, 155.32, 168.88; HRMS calcd for C16H25N3O2S m/z 324.1751 (M+ + H), found 324.1754.(S,Z)-tert-Butyl-1-(amino(methylthio)methyleneamino) propan-2-ylcarbamate, Boc-Ala-ψCH2-NH-C(SMe)-NH2 (3h)
Yield 97%; 1H NMR (300 MHz, CDCl3) δ 1.18 (d, J = 6.8 Hz, 3H), 1.39 (s, 9H), 2.08 (s, 3H), 3.45 (m, 2H), 3.83 (m, 1H), 5.14 (s, br, 1H), 5.86 (s, br, 1H); 13C NMR (750 MHz, CDCl3) δ 15.13, 18.17, 28.13, 45.45, 50.98, 80.34, 156.99, 168.54; HRMS calcd for C10H21N3O2S m/z 248.1431 (M+ + H), found 248.1434.(S)-Methyl-2-(3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)-3-methylbutyl)guanidino) propanoate, Fmoc-Val-ψ[CH2NHC(NH)NH]-Ala-OMe, (4a)
Yield 59%; 1H NMR (400 MHz, DMSO-d6) δ 0.97 (dd, J = 7.1 Hz, 6H), 1.40 (d, J = 7.2 Hz, 3H), 2.05–2.20 (m, 1H), 3.12 (m, 2H), 3.73 (s, 3H), 4.00–4.10 (m, 1H), 4.21 (t, J = 7.0 Hz, 1H), 4.25–4.50 (m, 2H), 4.50–4.65 (m, 1H), 5.56 (d, J = 8.8 Hz, 1H), 6.56 (s, br, 2H), 7.30 (m, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.59 (d, J = 7.1 Hz, 2H), 7.76 (d, J = 7.5 Hz, 2H); HRMS calcd for C25H34N4O4 m/z 453.0953 (M+ + H), found 453.0954.(S)-Methyl-3-(3-(2-(((9H-fluoren-9-yl)methoxy)carbonyl)-4-methylpentyl)guanidino) propanoate, Fmoc-Leu-ψ[CH2NHC(NH)NH]-β-Ala-OMe, (4b)
Yield 66%; 1H NMR (400 MHz, CDCl3) δ 0.92 (d, J = 6.8 Hz, 6H), 1.37 (m, 2H), 1.66 (m, 1H), 2.58 (m, 2H), 3.58 (s, 3H), 3.65–3.80 (m, 4H), 4.02 (m, 1H), 4.19 (t, J = 6.8 Hz, 1H), 4.35 (m, 2H), 5.21 (br, 1H), 6.96 (br, 2H), 7.22–7.76 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 22.44, 23.55, 25.29, 34.00, 42.35, 47.64, 52.31, 54.09, 64.32, 67.37, 120.48, 125.60, 127.61, 128.25, 141.75, 144.17, 157.77, 158.75, 173.44; HRMS calcd for C26H34N4O4 m/z 467.1827 (M+ + H), found 467.1894.(S)-Methyl-2-(3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)ethyl)guanidino)-3-phenylpropanoate, Fmoc-Gly-ψ[CH2NHC(NH)NH]-Phe-OMe, (4c)
Yield 63%; 1H NMR (400 MHz, CDCl3) δ 3.12–3.24 (m, 4H), 3.67 (s, 3H), 3.77–3.82 (m, 1H), 4.12–4.28 (m, 3H), 4.66 (d, J = 5.6 Hz, 2H), 5.17 (s, br, 1H), 6.45 (s, br, 2H), 7.15–7.41 (m, 9H), 7.53 (d, J = 7.2 Hz, 2H), 7.68 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 37.64, 39.81, 41.29, 47.69, 56.96, 60.71, 67.30, 120.44, 125.10, 125.65, 127.54, 128.15, 129.44, 129.83, 134.90, 141.78, 144.38, 156.16, 159.90, 173.89; HRMS calcd for C28H30N4O4 m/z 487.0804 (M+ + H), found 487.0806.(S)-Methyl-2-(3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)-2-phenylethyl)guanidino) propanoate, Fmoc-Phg-ψ[CH2-NHC(NH)NH]-(R)-Ala-OMe, (4e)
Yield 61%; 1H NMR (400 MHz, CDCl3) δ 1.17–1.18 (d, J = 4.0 Hz, 3H), 3.21–3.57 (m, 2H), 3.72 (s, 3H), 4.13–4.20 (m, 3H), 4.32 (d, J = 8.0 Hz, 2H), 5.24 (br, 1H), 6.10 (br, 1H), 7.28–7.48 (m, 8H), 7.56 (d, J = 4.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 17.12, 43.91, 46.31, 53.99, 58.26, 67.18, 120.03, 125.03, 127.09, 127.77, 128.33, 128.40, 128.63, 135.54, 141.30, 143.75, 155.14, 156.29, 171.17.(S)-Methyl-2-(3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)-2-phenylethyl)guanidino)propanoate, Fmoc-(R-)Phg-ψ[CH2-NHC(NH)NH]-(S)-Ala-OMe, (4e′)
Yield 60%; 1H NMR (400 MHz, CDCl3) δ 1.24–1.25 (d, J = 4.0 Hz, 3H), 3.20–3.51 (m, 2H), 3.74 (s, 3H), 4.10–4.19 (m, 3H), 4.32 (d, J = 8.0 Hz, 2H), 5.23 (br, 1H), 6.07 (br, 1H), 7.25–7.49 (m, 8H), 7.54 (d, J = 4.0 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H).(S)-Methyl-2-(3-((S)-2-(tert-butoxycarbonyl)-3-phenylpropyl)guanidino)-3-ethylbutanoate, Boc-Phe-ψ[CH2NHC(NH)NH]-Val-OMe, (4g)
Yield 71%; 1H NMR (400 MHz, DMSO-d6) 0.87 (d, J = 6.4 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 1.45 (s, 9H), 2.00–2.20 (m, 1H), 3.05–3.20 (m, 4H), 3.71 (s, 3H), 3.85–4.00 (m, 1H), 4.80–4.95 (m, 1H), 5.04 (d, J = 7.7 Hz, 1H), 6.37 (s, br, 2H), 7.05–7.15 (m, 2H), 7.25–7.35 (m, 3H); HRMS calcd for C21H34N4O4 m/z 407.2275 (M+ + H), found 407.2275.(S)-Ethyl-2-(3-((S)-2-(tert-butoxycarbonyl)propyl)guanidino)-3-methylbutanoate, Boc-Ala-ψ[CH2NHC(NH)NH]-Val-OEt, (4h)
Yield 68%; 1H NMR (400 MHz, CDCl3) δ 1.04 (m, 6H), 1.21 (d, J = 5.8 Hz, 3H), 1.41 (s, 9H), 1.96 (t, J = 6.9 Hz, 3H), 2.95 (m, 2H), 3.61 (m, 1H), 4.21 (m, 1H), 4.73 (m, 2H), 5.14 (s, br, 1H), 6.36 (br, 2H); 13C NMR (100 MHz, CDCl3) δ 16.92, 19.29, 21.47, 29.69, 30.22, 43.66, 48.24, 53.44, 65.23, 82.35, 158.07, 160.93, 172.93; HRMS calcd for C16H32N4O4 m/z 345.1477 (M+ + H), found 345.1472.(S)-Methyl-2-(((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)propylimino)methyleneamino)-3-methylbutanoate, Fmoc-Ala-ψ[CH2-NCN]-Val-OMe, (5a)
Yield 71%; IR (neat) νmax 2116 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.94 (dd, J = 6.8 Hz and J = 14.6 Hz, 6H), 1.34 (d, J = 7.2 Hz, 3H), 2.00–2.20 (m, 1H), 3.09 (m, 2H), 3.73 (s, 3H), 3.85–4.00 (m, 1H), 4.50–4.65 (m, 3H), 5.11 (d, J = 7.9 Hz, 1H), 7.03–7.60 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 19.59, 19.91, 20.60, 31.18, 44.59, 47.42, 52.47, 56.48, 62.60, 67.92, 120.54, 125.44, 125.52, 127.60, 128.33, 136.48, 141.75, 143.97, 157.24, 168.04; HRMS calcd for C25H29N3O4 m/z 436.5222 (M+ + H), found 436.5218.
(3S)-Benzyl-4-(2-benzyloxy)-2-oxoethylimino)methylene-amino)-3-benzyloxycarbonyl)butanoate, Cbz-Asp(OBzl)-ψ[CH2-NCN]-Gly-OBzl, (5b)
Yield 70%; IR (neat) νmax 2120 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.06–3.11 (m, 2H), 4.30 (m, 2H), 4.98 (s, 2H), 5.03 (s, 2H), 5.08 (s, 2H), 5.29 (s, 2H), 5.36 (m, 1H), 5.87 (d, J = 8.0 Hz, 1H), 7.18–7.28 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 44.39, 48.84, 55.14, 62.14, 67.66, 68.08, 68.84, 128.60, 128.86, 128.93, 129.04, 129.10, 129.13, 129.23, 129.35, 131.46, 132.68, 134.75, 135.56, 136.02, 156.28, 168.16, 170.53; HRMS calcd for C29H29N3O6 m/z 516.0697 (M+ + H), found 516.0665.
(4S)-Benzyl-4-(benzyloxycarbonyl)-5-((1-ethoxy-1-oxo-3-phenylpropan-2-ylimino)methyleneamino)pentanoate, Cbz-Glu(OBzl)-ψ[CH2-NCN]-Phe-OEt, (5c)
Yield 68%; IR (neat) νmax 2116 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.20 (t, J = 5.6 Hz, 3H), 1.82–1.94 (m, 2H), 2.13 (t, J = 4.5 Hz, 2H), 2.94–3.18 (m, 4H), 3.49 (m, 1H), 3.77 (m, 1H), 4.09–4.15 (m, 2H), 5.11 (s, 4H), 5.61 (s, br, 1H), 7.03–7.51 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 13.98, 28.01, 30.22, 37.79, 44.19, 50.01, 53.91, 61.47, 64.11, 67.18, 127.03, 127.95, 128.10, 128.17, 128.47, 129.21, 135.68, 136.13, 138.50, 155.27, 170.69, 173.01; HRMS calcd for C32H35N3O6 m/z 558.1796 (M+ + H), found 558.1702.
(S)-Methyl-2-(((S)-2-(benzyloxycarbonyl)-3-phenylpropylimino)methyleneamino)propanoate, Cbz-Phe-ψ[CH2-NCN]-Ala-OMe, (5d)
Yield 75%; IR (neat) νmax 2118 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.17 (d, J = 6.8 Hz, 3H), 3.11 (d, J = 4.8 Hz, 2H), 3.51 (m, 2H), 3.69 (s, 3H), 4.50 (t, J = 8 Hz, 1H), 5.04 (s, 2H), 5.22 (br, 1H), 5.66 (q, J = 8.4 Hz, 1H), 6.93–7.33 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 17.01, 34.05, 42.56, 50.78, 53.95, 57.61, 67.02, 126.52, 128.12, 128.26, 128.69, 128.83, 129.07, 136.85, 137.79, 139.94, 155.73, 173.56; HRMS calcd for C22H25N3O4 m/z 396.0901 (M+ + H), found 396.0908.
Benzyl(S)-phenyl-1-(((R)-1-phenylethylimino)methyleneamino)propan-2-ylcarbamate, Cbz-Phe-ψ[CH2-NCN]-R-phenethylamine, (5e)
Yield 74%; IR (neat) νmax 2119 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.48–1.49 (d, J = 6.0 Hz, 3H), 2.75 (m, 2H), 3.68 (m, 1H), 3.87 (m, 1H), 4.24 (m, 2H), 5.02 (s, 2H), 5.20 (br, 1H), 7.07–7.49 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 22.8, 38.5, 45.8, 52.3, 53.9, 66.8, 125.9, 126.8, 127.2, 127.7, 128.1, 128.5, 128.6, 128.9, 129.0, 136.2, 136.6, 139.8, 156.9, 179.0.
Benzyl(S)-phenyl-1-(((S)-1-phenylethylimino) methyleneamino)propan-2-ylcarbamate, Cbz-Phe-ψ[CH2-NCN]-S-phenethylamine, (5f)
Yield 73%; IR (neat) νmax 2120 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.54–1.55 (d, J = 6.0 Hz, 3H), 2.78 (m, 2H), 3.72 (m, 1H), 3.90 (m, 1H), 4.30 (m, 2H), 5.00 (s, 2H), 5.61 (br, 1H), 6.97–7.48 (m, 15H).
(S)-Methyl-2-(3-((S)-2-(benzyloxycarbonyl)-3-phenylpropyl) guanidino)propanoate, Cbz-Phe-ψ[CH2NHC(NH)NH]-Ala-OMe, (6a)
Yield 69%; 1H NMR (400 MHz, CDCl3) δ 1.36 (d, J = 7.1 Hz, 3H), 3.08 (m, 4H), 3.72 (s, 3H), 4.30–4.45 (m, 1H), 4.45–4.60 (m, 1H), 5.00 (s, 2H), 5.45 (s, br, 1H), 6.81 (br, 2H), 7.15–7.35 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 16.97, 33.65, 38.68, 47.32, 53.35, 60.73, 66.62, 126.60, 127.62, 127.94, 128.35, 128.50, 129.04, 136.14, 136.97, 156.79, 158.60, 172.54. HRMS calcd for C22H28N4O4 m/z 413.1994 (M+ + H), found 413.1974.(S)-Methyl-2-(3-((S)-2-(benzyloxycarbonyl)-3-methylbutyl)guanidino)-4-methylpentanoate, Cbz-Val-ψ[CH2NHC(NH)NH]-Leu-OMe, (6b)
Yield 67%; 1H NMR (400 MHz, CDCl3) 0.91 (d, J = 5.9 Hz, 6H), 0.93 (dd, J = 6.8 Hz and J = 12.6 Hz, 6H), 1.45–1.75 (m, 3H), 2.00–2.20 (m, 1H), 3.71 (s, 3H), 3.89 (m, 1H), 4.55–4.65 (m, 1H), 5.02 (s, 2H), 5.51 (s, br, 1H), 6.62 (br, 2H), 7.05–7.32 (m, 5H); HRMS calcd for C21H34N4O4 m/z 407.0785 (M+ + H), found 407.0781.(S)-Methyl-2-(((3-((S)-2-(benzyloxycarbonyl)-3-benzylthio)propyl)guanidino)propanoate, Cbz-Cys(Bzl)-ψ[CH2NHC(NH)NH]-Ala-OMe, (6c)
Yield 59%; 1H NMR (400 MHz, CDCl3) δ 1.32 (d, J = 7.2 Hz, 3H), 2.58 (m, 2H), 3.65 (s, 2H), 3.77 (s, 3H), 3.92–3.99 (m, 3H), 4.35 (m, 1H), 5.06 (s, 2H), 5.28 (d, J = 7.6 Hz, 1H), 6.84 (br, 2H), 7.23–7.32 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 16.76, 34.14, 36.91, 44.62, 49.61, 55.41, 55.56, 67.26, 127.69, 128.43, 128.63, 129.11, 129.56, 136.95, 138.07, 156.70, 158.80, 172.72; HRMS calcd for C23H30N4O4S m/z 459.1534 (M+ + H), found 459.1532.(S)-Methyl-3-(3-(2-(((9H-fluoren-9-yl)methoxy)carbonyl)propyl)guanidino)propanoate, Fmoc-Ala-ψ[CH2NHC(NH)NH]-β-Ala-OMe, (6d)
Yield 63%; 1H NMR (400 MHz, CDCl3) δ 1.12 (d, J = 6.8 Hz, 3H), 2.52 (br, 2H), 3.51 (s, 3H), 3.69 (br, 4H), 4.10 (m, 1H), 4.26 (t, J = 6.8 Hz, 2H), 4.45 (d, J = 6.2 Hz, 1H), 5.36 (br, 1H), 6.92 (br, 2H), 7.18–7.32 (m, 4H), 7.50 (d, J = 7.2 Hz, 2H), 7.67 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 19.15, 34.02, 40.26, 47.59, 47.95, 51.15, 52.34, 67.44, 120.48, 125.57, 127.59, 128.25, 141.75, 144.14, 157.46, 159.44, 173.46; HRMS calcd for C23H28N4O4 m/z 425.0537 (M+ + H), found 425.0530.(S)-Methyl-3-(3-(2-(((9H-fluoren-9-yl)methoxy)carbonyl)-3-phenylpropyl)guanidino)propanoate, Fmoc-Phe-ψ[CH2NHC(NH)NH]-β-Ala-OMe, (6e)
Yield 58%; 1H NMR (400 MHz, DMSO-d6) δ 2.56 (d, J = 6.4 Hz, 2H), 2.85 (m, 2H), 3.56 (s, 3H), 3.73 (m, 4H), 3.99 (br, 1H), 4.13 (t, J = 6.8 Hz, 1H), 4.22–4.34 (m, 2H), 5.45 (br, 1H), 6.78 (br, 2H), 7.18–7.51 (m, 13H); 13C NMR (100 MHz, DMSO-d6) δ 23.60, 30.19, 32.57, 33.99, 39.41, 47.54, 52.37, 67.45, 120.48, 125.58, 127.37, 127.60, 128.25, 129.23, 129.65, 137.50, 141.74, 144.21, 157.51, 159.53, 173.61; HRMS calcd for C29H32N4O4 m/z 501.1528 (M+ + H), found 501.1522.(S)-Methyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)propyl)-2-hydroxyguanidino)-3-methylbutanoate, Fmoc-Ala-ψ[CH2NHC(NOH)NH]-Val-OMe, (7a)
Yield 56%; 1H NMR (300 MHz, CDCl3) δ 0.98 (d, J = 4.8 Hz, 6H), 1.14 (d, J = 6.4 Hz, 3H), 2.64 (m, 3H), 3.56 (d, J = 7.1 Hz, 1H), 3.69 (s, 3H), 3.94 (m, 1H), 4.41 (t, J = 4.2 Hz, 1H), 4.67 (d, J = 5.6 Hz, 2H), 5.78 (br, 1H), 6.41 (br, 1H), 7.24–7.71 (m, 8H), 8.50 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 19.5, 19.9, 20.6, 30.1, 31.1, 40.5, 42.4, 47.4, 53.4, 66.6, 67.9, 120.5, 125.4, 125.5, 127.6, 128.3, 141.7, 143.9, 156.0, 159.2, 168.0; HRMS calcd for C25H32N4O5 m/z 491.1249 (M+ + Na), found 491.1240.(S)-Methyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)-3-phenylpropyl)-2-hydroxyguanidino)-3-phenyl propanoate, Fmoc-Phe-ψ[CH2NHC(NOH)NH]-Phe-OMe, (7b)
Yield 53%; 1H NMR (300 MHz, CDCl3) δ 2.72 (m, 1H), 2.96 (m, 4H), 3.14 (m, 1H), 3.69 (s, 3H), 3.78 (t, J = 7.0 Hz, 1H), 3.89 (t, J = 8.8 Hz, 1H), 4.46 (t, J = 6.2 Hz, 1H), 4.68 (d, J = 5.9 Hz, 2H), 5.48 (br, 1H), 6.68 (br, 1H), 7.17–7.81 (m, 18H), 8.46 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 35.8, 39.6, 42.0, 46.9, 53.4, 56.4, 66.7, 68.3, 126.3, 126.4, 126.6, 127.7, 127.8, 127.9, 128.0, 128.6, 128.7, 128.9, 138.4, 138.6, 141.0, 143.1, 156.0, 160.3, 170.8; HRMS calcd for C35H36N4O5 m/z 615.1477 (M+ + Na), found 615.1472.(S,E)-Ethyl-2-(3-(2-(((9H-fluoren-9-yl)methoxy)carbonyl)-3-phenylpropyl)-2-cyanoguanidino)acetate, Fmoc-Phe-ψ[CH2NHC(NCN)NH]-Gly-OEt, (8a)
Yield 64%; 1H NMR (300 MHz, CDCl3) δ 1.21 (t, J = 4.7 Hz, 3H), 2.59 (d, J = 3.2 Hz, 1H), 2.67 (d, J = 5.6 Hz, 1H), 2.75 (d, J = 5.7 Hz, 2H), 3.49 (s, 2H), 3.85 (m, 1H), 4.05 (m, 2H), 4.32 (t, J = 5.9 Hz, 1H), 4.65 (d, J = 3.1 Hz, 2H), 5.48 (s, br, 1H), 6.10 (s, br, 1H), 6.82 (s, br, 1H), 7.15–7.79 (m, 13H); 13C NMR (75 MHz, CDCl3) δ 14.1, 38.4, 41.8, 47.6, 50.0, 60.2, 67.5, 116.5, 125.4, 127.0, 127.6, 128.2, 129.1, 129.6, 130.5, 130.9, 137.9, 141.8, 144.1, 153.9, 156.0, 170.1; HRMS calcd for C30H31N5O4 m/z 548.0785 (M+ + Na), found 548.0781.(S)-Benzyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)-3-phenylpropyl)-2-cyanoguanidino)-4-methyl pentanoate, Fmoc-Phe-ψ[CH2NHC(NCN)NH]-Leu-OBzl, (8b)
Yield 67%; 1H NMR (300 MHz, CDCl3) δ 0.82 (d, J = 5.4 Hz, 6H), 1.42 (t, J = 7.1 Hz, 2H), 1.49–1.63 (m, 1H), 2.51 (d, J = 3.9 Hz, 1H), 2.59 (d, J = 6.5 Hz, 1H), 2.65 (d, J = 6.2 Hz, 2H), 3.29 (t, J = 11.1 Hz, 1H), 3.89 (m, 1H), 4.36 (t, J = 4.9 Hz, 1H), 4.74 (d, J = 3.9 Hz, 2H), 5.22 (s, 2H), 6.01 (s, br, 1H), 6.39 (s, br, 1H), 6.92–7.75 (m, 18H and NH); 13C NMR (75 MHz, CDCl3) δ 22.8., 23.0, 34.3, 40.4, 42.2, 47.4, 48.0, 60.03, 65.3, 67.7, 117.5, 125.7, 126.4, 127.6, 128.2, 128.6, 128.9, 129.2, 129.8, 136.6, 139.7, 141.7, 143.1, 154.4, 156.2, 171.1; HRMS calcd for C35H33N5O4 m/z 610.1067 (M+ + Na), found 610.1061.(S)-Methyl-2-((E)-3-((S)-2-(benzyloxycarbonyl)-4-methylpentyl)-2-aminoguanidino)-3-phenylpropanoate, Z-Leu-ψ[CH2NHC(NNH2)NH]-Phe-OMe, (9a)
Yield 71%; 1H NMR (300 MHz, CDCl3) δ 0.98 (d, J = 6.8 Hz, 6H), 1.42 (m, 2H), 1.91 (m, 1H), 2.79 (m, 2H), 3.12 (m, 1H), 3.26 (m, 1H), 3.71 (s, 3H), 4.38 (m, 1H), 4.92 (m, 1H), 5.04 (s, 2H), 6.08 (br, 1H), 6.72 (br, 1H), 7.12–7.22 (m, 10H), 7.84 (br, 2H); 13C NMR (75 MHz, CDCl3) δ 18.6, 21.2, 36.3, 43.2, 45.1, 52.3, 56.7, 64.8, 67.9, 127.1, 127.8, 128.2, 128.6, 129.4, 129.7, 137.2, 138.2, 155.4, 156.1, 170.1; HRMS calcd for C25H35N5O4 m/z 470.0275 (M+ + H), found 470.0275.(S)-Methyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)propyl)-2-aminoguanidino)-3-methylbutanoate, Fmoc-Ala-ψ[CH2NHC(NNH2)NH]-Val-OMe, (9b)
Yield 51%; 1H NMR (300 MHz, CDCl3) δ 0.84 (d, J = 6.4 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 1.65 (d, J = 6.8 Hz, 3H), 2.81 (m, 3H), 3.52 (s, 3H), 4.23 (m, 1H), 4.36 (m, 1H), 4.99 (d, J = 8.4 Hz, 2H), 5.16 (m, 1H), 5.49 (br, 1H), 6.76 (br, 1H), 7.21 (m, 2H), 7.32 (m, 2H), 7.45 (d, 2H, J = 6.8 Hz), 7.67 (d, 2H, J = 7.6 Hz), 8.07 (br, 2H); 13C NMR (75 MHz, CDCl3) δ 16.80, 21.6, 29.8, 44.2, 46.8, 52.4, 59.6, 63.2, 65.8, 126.7, 127.2, 127.9, 128.4, 141.1, 142.8, 154.1, 156.0, 170.4; HRMS calcd for C25H33N5O4 m/z 468.0642 (M+ + H), found 468.0649.(2S,3R)-methyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)propyl)-2-phenylguanidino)-3-methylpentanoate, Fmoc-Ala-ψ[CH2NHC(NC6H5)NH]-Ile-OMe, (10a)
Yield 65%; 1H NMR (300 MHz, CDCl3) δ 0.94 (t, J = 4.2 Hz, 3H), 0.99 (d, J = 4.6 Hz, 3H), 1.24 (m, 2H), 1.29 (d, J = 6.8 Hz, 3H), 2.04 (m, 1H), 2.94 (m, 2H), 3.71 (s, 3H), 4.02 (m, 1H), 4.42 (t, J = 6.1 Hz, 1H), 4.62 (d, J = 7.1 Hz, 2H), 4.89 (m, 1H), 5.41 (br, 1H), 6.12 (br, 1H), 6.73 (br, 1H), 7.10–7.75 (m, 13H); 13C NMR (75 MHz, CDCl3) δ 10.4, 16.3, 18.9, 28.6, 35.0, 42.4, 46.4, 48.2, 50.6, 52.3, 66.4, 120.6, 125.4, 125.8, 127.4, 127.6, 128.4, 128.8, 139.1, 141.0, 143.2, 155.2, 155.9, 172.1; HRMS calcd for C32H38N4O4 m/z 565.1796 (M+ + Na), found 565.1702.(S,E)-methyl-2-(3-(2-(((9H-fluoren-9-yl)methoxy)carbonyl)ethyl)-2-benzylguanidino)-4-methylpentanoate, Fmoc-Gly-ψ[CH2NHC(NBn)NH]-Leu-OMe, (10b)
Yield 58%; 1H NMR (300 MHz, CDCl3) δ 0.92 (d, J = 5.4 Hz, 6H), 1.45–1.71 (m, 3H), 2.52–2.63 (m, 2H), 2.79 (m, 2H), 2.97 (s, 2H), 3.31 (t, J = 4.2 Hz, 1H), 3.69 (s, 3H), 4.32 (t, J = 2.9 Hz, 1H), 4.49 (d, J = 7.2 Hz, 2H), 5.69 (s, br, 1H), 6.24 (s, br, 1H), 6.75 (s, br, 1H), 7.09–7.81 (m, 13H), 13C NMR (75 MHz, CDCl3) δ 21.8, 22.5, 38.4, 41.1, 44.2, 46.9, 50.8, 52.0, 54.3, 67.2, 119.8, 125.0, 126.8, 126.9, 127.6, 128.3, 129.2, 136.2, 141.1, 143.7, 154.3, 156.6, 170.7; HRMS calcd for C32H38N4O4 m/z 565.1615 (M+ + Na), found 565.1603.(S)-Methyl-2-((E)-3-((S)-2-(((9H-fluoren-9-yl)methoxy)carbonyl)propyl)-2-(3-chlorophenyl)guanidino)-3-methylbutanoate, Fmoc-Ala-ψ[CH2NHC(NC6H4Cl)NH]-Val-OMe, (10c)
Yield 62%; 1H NMR (300 MHz, CDCl3) δ 0.89 (d, J = 3.7 Hz, 6H), 1.17 (d, J = 6.4 Hz, 3H), 2.12 (m, 1H), 2.41 (d, J = 4.8 Hz, 1H), 2.82 (d, J = 7.1 Hz, 1H), 2.89 (d, J = 11.2 Hz, 1H), 3.52 (s, 3H), 3.85 (m, 1H), 4.51 (t, J = 6.6 Hz, 1H), 4.81 (d, J = 5.6 Hz, 2H), 6.12 (s, br, 1H), 6.93 (s, br, 1H), 7.12–7.75 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 18.0, 18.9, 30.9, 42.7, 46.9, 49.4, 55.4, 57.9, 66.6, 119.7, 124.9, 126.8, 127.5, 128.0, 128.3, 135.4, 141.0, 143.6, 143.7, 153.9, 156.6, 170.1,; ES-MS calcd for C31H35ClN4O4 m/z 586.09 (M+ + Na), found 586.07.Acknowledgements
We thank Professor K. N. Ganesh, IISER Pune, for useful discussions at the beginning of this study. Also we thank DST, and BRNS for their generous financial assistance. Authors also thank Dr H. N. Gopi, IISER, Pune for Mass spectrometry and SAIF, IPC, IISc, Bangalore for 1H and 13C NMR spectra. One of the authors Basavaprabhu thanks CSIR for SRF fellowship.Notes and references
- (a) P. S. Farmer, in Drug Design, ed. E. J. Ariens, Academic Press, San Diego, 1980, vol. 10, p. 119 Search PubMed; (b) A. F. Spatola, in Chemistry and Biochemistry of Amino Acids, Peptides, and Proteins: A Survey of Recent Developments, ed. B. Weinstein, Marcel Dekker, New York, 1983, vol. 7, p. 267 Search PubMed; (c) K. S. Lam, M. Lebl and V. Krchnak, Chem. Rev., 1997, 97, 411 CrossRef CAS PubMed; (d) J.-M. Ahn, N. A. Boyle, M. T. MacDonald and K. D. Janda, Mini-Rev. Med. Chem., 2002, 2, 463 CrossRef CAS PubMed.
- (a) M. Goodman, Synthesis of Peptides and Peptidomimetics, Houben-Weyl E22c, Thieme, 2002 Search PubMed; (b) L. Wang and O. Phanstiel, IV, J. Org. Chem., 2000, 65, 1442 CrossRef CAS PubMed; (c) R. A. Breitenmoser, A. Linden and H. Heimgartner, Helv. Chim. Acta, 2002, 85, 990 CrossRef CAS; (d) T. M. Vishwanatha, N. Narendra, B. Chattopadhyay, M. Mukherjee and V. V. Sureshbabu, J. Org. Chem., 2012, 77, 2689 CrossRef CAS PubMed; (e) Y. L. Angel and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674 RSC; (f) E. Mann and H. Kessler, Org. Lett., 2003, 5, 4567 CrossRef CAS PubMed.
- (a) B. S. Patil, G.-R. Vasanthkumar and V. V. Sureshbabu, J. Org. Chem., 2003, 68, 7274 CrossRef CAS PubMed; (b) V. V. Sureshbabu, B. S. Patil and R. Venkataramanarao, J. Org. Chem., 2006, 71, 7697 CrossRef CAS PubMed.
- V. V. Sureshbabu, S. A. Naik, N. Narendra and H. P. Hemantha, J. Org. Chem., 2009, 74, 5260 CrossRef CAS PubMed.
- G. Chennakrishnareddy, G. Nagendra, H. P. Hemantha and V. V. Sureshbabu, Tetrahedron, 2010, 66, 6718 CrossRef CAS.
- (a) R. G. S. Berlinck and M. H. Kossuga, Nat. Prod. Rep., 2005, 22, 516 RSC; (b) R. G. S. Berlinck, A. C. B. Burtoloso and M. H. Kossuga, Nat. Prod. Rep., 2008, 25, 919 RSC.
- T. Suhs and B. Konig, Chem.–Eur. J., 2006, 12, 8150 CrossRef CAS PubMed.
- (a) Z. Zhang and E. Fan, J. Org. Chem., 2005, 70, 8801 CrossRef CAS PubMed; (b) Z. Zhang, T. Carter and E. Fan, Tetrahedron Lett., 2003, 44, 3063 CrossRef CAS.
- (a) J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292 CrossRef CAS PubMed; (b) J. A. Patch and A. E. Barron, Curr. Opin. Chem. Biol., 2002, 6, 872 CrossRef CAS PubMed and references cited therein.
- For reviews on the synthesis of guanidines in solution see: (a) T. Suhs and B. Konig, Mini-Rev. Med. Chem., 2006, 3, 315 CrossRef CAS and references cited there in; (b) A. R. Katritzky and B. V. Rogovoy, ARKIVOC, 2005, iv, 49 Search PubMed; (c) T. Ishikawa and T. Kumamoto, Synthesis, 2006, 737 CrossRef CAS; (d) R. G. S. Berlinck, M. H. Rossuga and A. M. Nascimento, Sci. Synth., 2005, 18, 1117 Search PubMed; (e) T. Ishikawa, ARKIVOC, 2006, 7, 148 Search PubMed.
- Note: For the solid phase synthesis of guanidines see: (a) S. Sandanayake, S. Perera and N. J. Ede, QSAR Comb. Sci., 2004, 23, 655 CrossRef CAS; (b) J. P. Kilburn, J. Lau and R. C. F. Jones, Tetrahedron, 2002, 58, 1739 CrossRef CASL. Gomez, F. Gellibert, A. Wagner and C. Mioskowski, Chem.–Eur. J., 2000, 6, 4016 CrossRef CAS and references cited therein.
- (a) B. Rathke, Chem. Ber., 1884, 17, 297 CrossRef; (b) M. Bonnat, M. Bradley and J. D. Kilbum, Tetrahedron Lett., 1996, 37, 5409 CrossRef CAS.
- (a) V. V. Sureshbabu, T. M. Vishwanatha and B. Vasantha, Synlett, 2010, 1037 CrossRef CAS; (b) R. Pathania, S. Zlitni, C. Barker, R. Das, D. A. Gerritsma, J. Lebert, J. E. Awuah, G. Melacini, F. A. Capretta and E. D. Brown, Nat. Chem. Biol., 2009, 5, 849 CrossRef CAS PubMed.
- (a) G. Kjellin and J. Sandstrom, Acta Chem. Scand., 1969, 8, 23 Search PubMed; (b) G. R. Clemo, T. Holmes and G. C. Leitch, J. Chem. Soc., 1938, 753 RSC; (c) R. L. Grant and F. L. Pyman, J. Chem. Soc., 1921, 119, 1893 RSCR. J. Herr, L. J. Kuhler, H. Meckler and C. J. Opalka, Synthesis, 2000, 1569 CrossRef CAS.
- C. R. Rasmussen, F. J. Villani, Jr, L. E. Weaner, B. E. Reynolds, A. R. Hood, L. R. Hecker, S. O. Nortey, A. Hanslin, M. J. Costanzo, E. T. Powell and A. J. Molinari, Synthesis, 1988, 456 CrossRef CAS.
- J. Stadlwieser, E. P. Ellmerer-Muller, A. Tako, N. Maslouh and W. Bannwarth, Angew. Chem., Int. Ed., 1998, 37, 1402 CrossRef CAS.
- (a) A. R. Katritzky, N. Kirichenko, B. V. Rogovoy, J. Kister and H. Tao, Synthesis, 2004, 1799 CrossRef CAS; (b) P. L. Feldman, Tetrahedron Lett., 1991, 32, 875 CrossRef CAS.
- Note: Chiral HPLC profiles of the two isomers, 4e and 4e′ had peaks at Rt values 8.45 and 9.75 min respectively and the equimolar mixture of the intentionally made epimers showed two well separated peaks corresponding to the guanidinopeptide mimics at Rt = 8.45 and 9.73 min. Thus, from these studies it is evident that the samples analyzed were optically pure and the coupling of isothiouronium derivatives with amino acid esters took place with retention of configuration at the chiral center of the isothiouronium derivatives as well as that of the newly coupled amino acid ester. (Method: λ = 254 nm; flow rate: 1.0 mL min−1; chiralpak IA, 250 × 4.6 mm; 5 μL; isocratic: hexane: EtOH; 9:1). See ESI† for chromatograms.
- For biological applications of N-hydroxy guanidine see: (a) R. A. Pufahl, P. G. Nanjappan, R. W. Woodard and M. A. Marletta, Biochemistry, 1992, 31, 6822 CrossRef CAS PubMed; (b) M. Xian, N. Fujiwara, Z. Wen, T. Cai, S. Kazuma, A. J. Janczuk, X. Tang, V. V. Telyatnikov, Y. Zhang, X. Chen, Y. Miyamato, N. Taniguchi and P. G. Wang, Bioorg. Med. Chem., 2002, 10, 3049 CrossRef CAS PubMed; (c) T. Cai, M. Xian and P. G. Wang, Bioorg. Med. Chem. Lett., 2002, 12, 1507 CrossRef CAS PubMed; (d) R. Ricoux, J.-L. Boucher, D. Mandon, Y. M. Frapart, Y. Henry, D. Mansuy and J.-P. Mahy, Eur. J. Biochem., 2003, 270, 47 CrossRef CAS PubMed.
- For the applications of aminoguanidine see: E. Lieber and G. B. L. Smith, Chem. Rev., 1938, 38, 213 Search PubMed.
- For the applications of cyanoguanidine see: L. A. Sheludyakova, E. V. Sobolev, A. V. Arbuznikov, E. B. Burgina and L. I. Kozhevina, J. Chem. Soc., Faraday Trans., 1997, 93, 1357 RSC.
- P. S. Dangate and K. G. Akamanchi, Tetrahedron Lett., 2012, 53, 6765 CrossRef CAS and references cited therein.
- J. I. Gavrilyuk, G. Evindar and R. A. Batey, J. Comb. Chem., 2006, 8, 237 CrossRef CAS PubMed.
- J. I. Gavrilyuk, G. Evindar, J. Y. Chen and R. A. Batey, J. Comb. Chem., 2007, 9, 644 CrossRef CAS PubMed.
- M. Heras, M. Ventura, A. Linden and J. M. Villalgordo, Tetrahedron, 2001, 57, 4371 CrossRef CAS.
- J. H. Kang, J. T. Moon, J. Kim, D. J. Joo and J. Y. Lee, Bull. Korean Chem. Soc., 2007, 28, 913 CrossRef CAS.
- S. Bepary, I. K. Youn, H.-J. Lim and G. H. Lee, Eur. J. Org. Chem., 2012, 2542 CrossRef CAS.
- F. Palacios, C. Alonso, D. Aparicio, G. Rubiales and J. M. de Los Santos, Tetrahedron, 2007, 63, 523 CrossRef CAS and references cited therein.
- (a) N. Narendra, T. M. Vishwanatha and V. V. Sureshbabu, Synthesis, 2011, 3247 CAS; (b) R. S. Lamani, G. Nagendra and V. V. Sureshbabu, Tetrahedron Lett., 2010, 51, 4705 CrossRef CAS.
- G. D. Meakin and R. J. Moss, J. Chem. Soc., 1957, 993 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of mass spectrometry, 1H and 13C NMR spectra of compounds prepared were given in ESI. See DOI: 10.1039/c4ra07252a |
| This journal is © The Royal Society of Chemistry 2014 |