
Phosphate tricyclic coumarin analogs as steroid sulfatase inhibitors: synthesis and biological activity†
Abstract
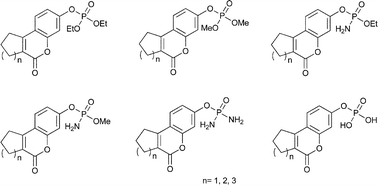
In the present work, we report convenient methods for the synthesis and biological evaluation of phosphate tricyclic coumarin derivatives as potential steroid sulfatase inhibitors. The described synthesis includes the straightforward preparation of 7-hydroxy-2,3-dihydro-1H-cyclopenta[c]chromen-4-one, 3-hydroxy-7,8,9,10-tetrahydro-6H-benzo[c]chromen-6-one and 3-hydroxy-8,9,10,11-tetrahydro-7H-cyclohepta[c]chromen-6-one modified with various phosphate moieties. The inhibitory effects of the synthesized compounds were tested on STS isolated from human placenta as well as the MCF-7, MDA-MB-231 and MDA-MB-435S cancer cell lines. Most of the new STS inhibitors possessed IC50 values between 21 to 159 μM. In the course of our investigation, the largest inhibitory effects in the STS enzyme assays were observed for the three compounds 9p, 9r and 9s, with IC50 values of 36.4, 37.8 and 21.5 μM, respectively (IC50 value of 1.0 μM for the 665-COUMATE used as a reference). The compound 9r, exhibited the highest potency against MCF-7, an estrogen receptor positive (ER+) cell line, with a GI50 value of 24.7 μM. The structure-activity relationships of the synthesized coumarin derivatives with the STS enzyme are discussed.
 Please wait while we load your content...
Please wait while we load your content...

