
Crystallization behaviour of a polymeric membrane based on the polymer PVdF–HFP and the ionic liquid BMIMBF4†
Abstract
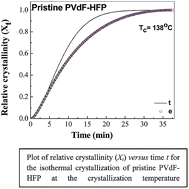
The crystallization behaviour of the polymer poly(vinylidenefluoride)–hexafluoropropylene (PVdF–HFP) in the presence and absence of the ionic liquid (IL) (1-butyl-3-methylimidazolium tetrafluoroborate; [BMIMBF4]) were studied by isothermal and non-isothermal crystallization processes using differential scanning calorimetry. The well-known Avrami equation is used to describe the isothermal crystallization process of pristine PVdF–HFP or PVdF–HFP + x wt% of IL BMIMBF4, where x = 10 and 30, respectively. It was found that the presence of the IL BMIMBF4 in the PVdF–HFP matrix suppresses the crystallization of the polymer PVdF–HFP, resulting in low crystal growth rates. Three kinetic methods (i.e., those of Jeziorny, Ozawa and Mo) were used to analyze the non-isothermal crystallization process. The Avrami equation modified by Jeziorny could only describe the initial stage of crystallization and the Ozawa method failed to describe the non-isothermal crystallization behavior, but Mo's method explains the results better.
 Please wait while we load your content...
Please wait while we load your content...

