
pH-responsive luminescence of a new trinuclear Ru(II) polypyridine complex†
A. Arun Viveke and
V. Alexander*
Department of chemistry, Loyola College, Chennai, Tamilnadu, India. E-mail: valexander@rediffmail.com
First published on 8th September 2014
Abstract
Synthesis of a new heteroleptic trinuclear Ru(II) complex of a ditopic imidazole-based terpyridine bridging ligand and its room temperature luminescence in fluid solution and pH modulated luminescence behaviour is reported.
Ruthenium(II) complexes of polypyridine ligands continue to be an active area of research owing to their excellent spectroscopic and photochemical properties and their use in photocatalysis,1 dye sensitised solar cells,2 molecular electronics,3 molecular machines and motors,4 metallodendrimers,5 light-to-chemical energy conversion schemes,6 as light harvesting antennas,7 DNA intercalaters,8 and building blocks for macromolecular assemblies that are of interest in biochemistry and chemical diagnosis.9 Multimetallic supramolecular assemblies constructed from ruthenium polypyridine chromophores have been extensively investigated because they exhibit intercomponent energy and/or electron transfer processes, possibly leading to valuable functions such as charge separation and/or energy migration.10 When the spacer linking the coordinating sites of the bridging ligand is not rigid, the geometry of the assembly is not defined. Such systems are of limited applicative interest because practical devices usually require the occurrence of vectorial energy- or electron transfer over long distance. Therefore, rigid spacers are preferred.11
The imidazole-containing ligands are poor π-acceptors and better π-donors and control orbital energies by proton transfer.12 Ru(II) complexes of imidazole containing ligands where the imidazole rings are coordinated to the metal ion are usually nonemissive or weakly emissive in fluid solution at room temperature, while those with uncoordinated imidazole rings are good emitters with proton induced “on–off” emission switching characteristics.12 Structurally appealing [Ru(tpy)2]2+ (tpy = 2,2′:6′,2′′-terpyridine) and photophysically fascinating [Ru(bpy)3]2+ are the widely investigated prototypes of ruthenium polypyridine family. Though their photophysics and electrochemistry are extensively investigated, complexes in which [Ru(tpy)]2+ and [Ru(bpy)]2+ units are connected through rigid conjugated bridges are rare. We report the synthesis, photophysical and electrochemical properties and pH modulation of the photophysical behaviour of the homotrinuclear Ru(II) complex 3 based on the ditopic imidazole-based terpyridine bridging ligand ttpy-Izphen (ttpy-Izphen = 2-(4-(2,6-di(pyridin-2-yl)pyridin-4-yl)-phenyl)-1H-imidazo[4,5-f][1,10]phenanthroline). When our study was in progress Baitalik et al.13 have reported the homo- and heterodinuclear (Ru–Ru and Ru–Rh) complexes of 1. They reported luminescence quenching in the homodinuclear Ru(II) complexes with terpyridine and tolylterpyridine auxiliary ligands. In the present study, we observe luminescence from the homotrinuclear Ru(II) complex 3 at room temperature in fluid solution and pH modulated luminescence characteristics.
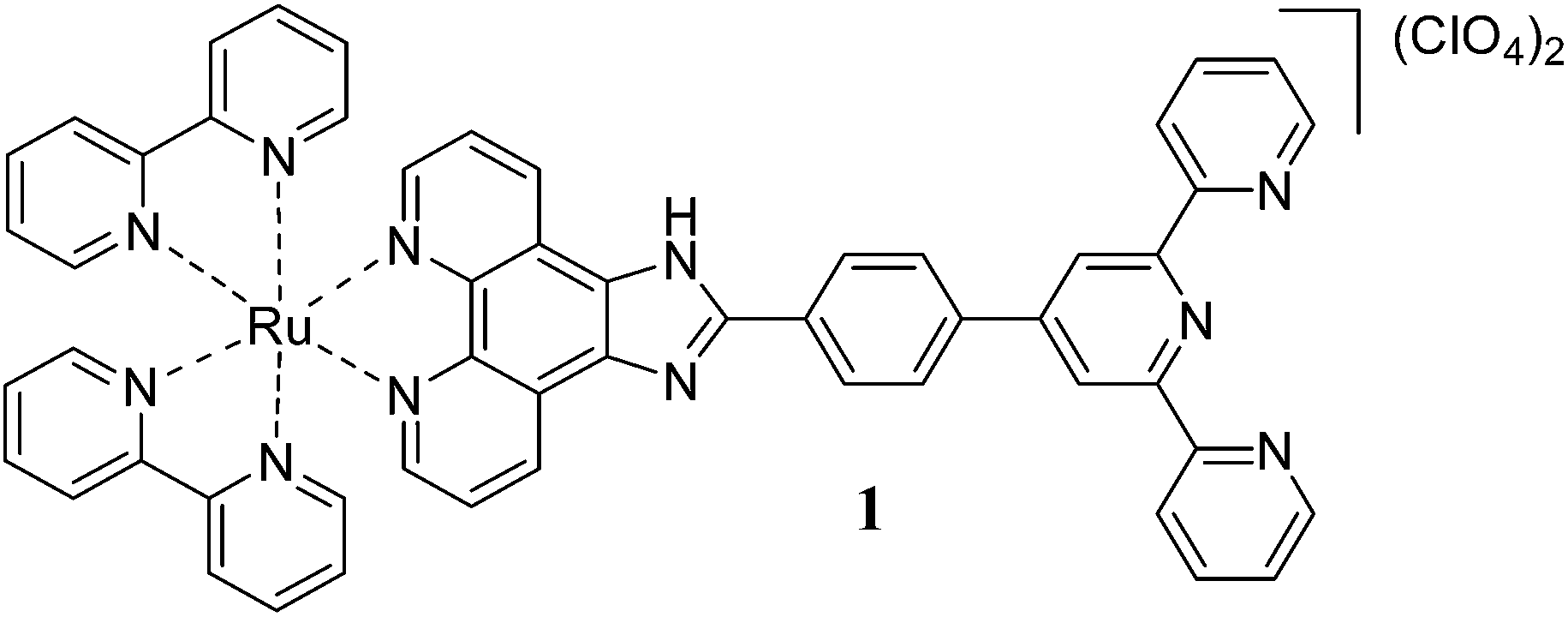
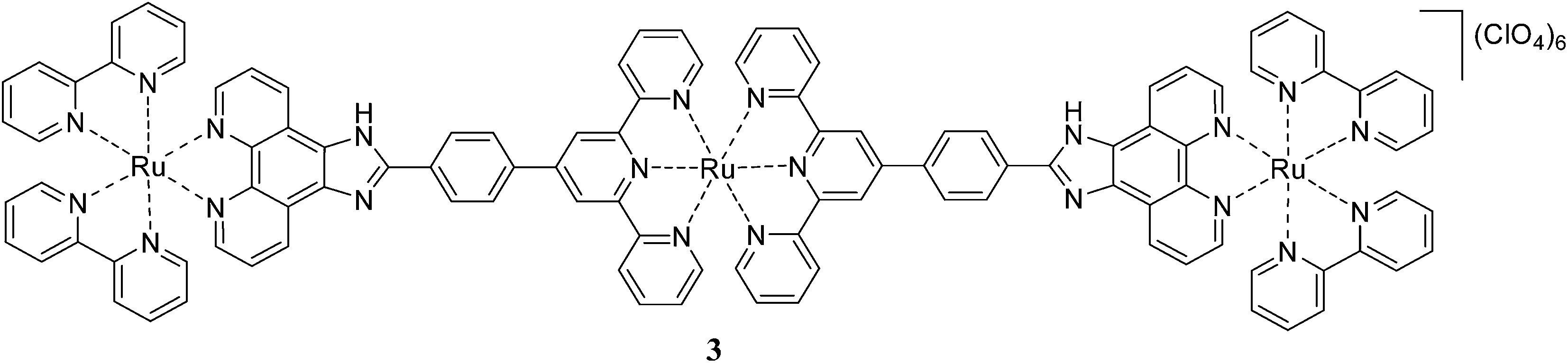
The homotrinuclear Ru(II) complex 3 is synthesized by a two stage strategy via the synthesis of the dinuclear intermediate complex 2, synthesised by the reaction of 1 with the RuCl3·H2O, followed by its reaction with 1. Complexes 1 and 3 are characterised by IR, ESI mass spectrometry, 1H NMR, and 1H–1H COSY NMR spectroscopy.
The mononuclear Ru(II) complex 1 exhibits electronic absorption bands at 286 and 333 nm assignable to the π → π* transitions of the ligand and the Rudπ → bpy and Rudπ → ttpy-Izphen 1MLCT transitions at 429 and 458 nm. The trinuclear Ru(II) complex 3 exhibits electronic absorption bands at 286 and 334 nm assignable to the π → π* transitions of the ligand and a shoulder at 356 nm corresponding to the metal centered absorption arising out of the coordination of the terpyridine moiety.14 The absorption bands at 432 and 466 nm are due to the Rudπ → bpy and Rudπ → ttpy-Izphen 1MLCT transitions and the band at 496 nm is characteristic of bis(terpyridine)ruthenium(II) complexes. The Rudπ → ttpy-Izphen 1MLCT transition occurs at lower energy than the Rudπ → bpy transition.15 The 1MLCT bands of 3 encounter hyperchromic shift when compared to that of 1.
The emission spectra of 1 and 3 are recorded in CH3CN at 298 K and CH3OH–C2H5OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v/v) rigid glass at 77 K. Upon excitation at the excitation maxima the mononuclear complex 1 emits from the 3MLCT state at 606 nm at room temperature. The luminescence spectrum of 1 is reported earlier by different groups13,15 and our results match with negligible variation in the emission wavelength maxima. The trinuclear Ru(II) complex 3 exhibits a broad emission band at 599 nm. It is interesting to note that the complex 3 exhibits room temperature luminescence unlike its dinuclear Ru(II) complexes [(bpy)2RuII(phen-Hbzim-tpy)RuII(tpy/ttpy)]4+ reported by Baitalik et al.13 The room temperature emission profile and emission maxima for 1 and 3 are independent of the excitation wavelength and the corrected excitation spectra match with the absorption spectra (Fig. S12, ESI†). The emission of the complex 3 at 599 nm indicates that it arises from the [(bpy)2Ru(ttpy-Izphen)] unit since the emission from the tpy-based Ru(II) chromophores usually occur at or above 640 nm. The luminescence quantum yield of 3 (0.111) is lower than that of 1 (0.290) which could be attributed to the presence of an excited state non-radiative process. The complex 3 exhibits a biexponential decay with a shorter lifetime component (τ = 9.70 ns) and a longer lifetime component (τ = 112 ns), while the mononuclear complex 1 shows a single exponential decay with a lifetime of 159 ns (decay profiles are presented in Fig. 1a). It is evident that the longer lifetime component of 3 is from the [(bpy)2Ru(ttpy-Izphen)] based excited state, while the shorter lifetime component may originate from the [Ru(ttpy-Izphen)2] based excited state. By comparing with the analogous Ru(II) polypyridine complexes, the longer lifetime of 3 is attributed to the radiative transition from the [(bpy)2Ru(ttpy-Izphen)] excited state and the shorter lifetime component should emerge from the [Ru(ttpy-Izphen)2] excited state.16 Thus, the role of the ditopic bridging ligand ttpy-Izphen in effectively stabilizing the 3MLCT state by increasing the energy gap between the 3MLCT and 3MC state is evident.17 In the case of the trinuclear complex 3 the ken for the energy transfer from the excited state of [(bpy)2Ru(ttpy-Izphen)] based component to the ground-state of [Ru(ttpy-Izphen)2] based component is lower (2.63 × 106 s−1) than that of the [(bpy)2RuII(phen-Hbzim-tpy)RuII(tpy/ttpy)]4+ systems (ken = 5.71 × 107/4.77 × 107 s−1)13 indicating that the lower value of ken may favour the room temperature luminescence of 3 (Table 1).
:4, v/v) rigid glass at 77 K. Upon excitation at the excitation maxima the mononuclear complex 1 emits from the 3MLCT state at 606 nm at room temperature. The luminescence spectrum of 1 is reported earlier by different groups13,15 and our results match with negligible variation in the emission wavelength maxima. The trinuclear Ru(II) complex 3 exhibits a broad emission band at 599 nm. It is interesting to note that the complex 3 exhibits room temperature luminescence unlike its dinuclear Ru(II) complexes [(bpy)2RuII(phen-Hbzim-tpy)RuII(tpy/ttpy)]4+ reported by Baitalik et al.13 The room temperature emission profile and emission maxima for 1 and 3 are independent of the excitation wavelength and the corrected excitation spectra match with the absorption spectra (Fig. S12, ESI†). The emission of the complex 3 at 599 nm indicates that it arises from the [(bpy)2Ru(ttpy-Izphen)] unit since the emission from the tpy-based Ru(II) chromophores usually occur at or above 640 nm. The luminescence quantum yield of 3 (0.111) is lower than that of 1 (0.290) which could be attributed to the presence of an excited state non-radiative process. The complex 3 exhibits a biexponential decay with a shorter lifetime component (τ = 9.70 ns) and a longer lifetime component (τ = 112 ns), while the mononuclear complex 1 shows a single exponential decay with a lifetime of 159 ns (decay profiles are presented in Fig. 1a). It is evident that the longer lifetime component of 3 is from the [(bpy)2Ru(ttpy-Izphen)] based excited state, while the shorter lifetime component may originate from the [Ru(ttpy-Izphen)2] based excited state. By comparing with the analogous Ru(II) polypyridine complexes, the longer lifetime of 3 is attributed to the radiative transition from the [(bpy)2Ru(ttpy-Izphen)] excited state and the shorter lifetime component should emerge from the [Ru(ttpy-Izphen)2] excited state.16 Thus, the role of the ditopic bridging ligand ttpy-Izphen in effectively stabilizing the 3MLCT state by increasing the energy gap between the 3MLCT and 3MC state is evident.17 In the case of the trinuclear complex 3 the ken for the energy transfer from the excited state of [(bpy)2Ru(ttpy-Izphen)] based component to the ground-state of [Ru(ttpy-Izphen)2] based component is lower (2.63 × 106 s−1) than that of the [(bpy)2RuII(phen-Hbzim-tpy)RuII(tpy/ttpy)]4+ systems (ken = 5.71 × 107/4.77 × 107 s−1)13 indicating that the lower value of ken may favour the room temperature luminescence of 3 (Table 1).
 | ||
| Fig. 1 (a) Decay profiles of 1 (black) and 3 (red); (b) emission spectrum of 3 in frozen CH3OH–C2H5OH (1:4, v/v) at 77 K (λex = 456 nm) (red line) and (λex = 496 nm) (dotted line). | ||
| Absorption λmax/nm (ε × 104/M−1 cm−1) | Luminescence | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 298 K | 77 K | ||||||||
| λmax/nm | τ/ns | Φb | krc/105 (s−1) | knrd 107 (s−1) | kene (s−1) | λmax/nm | Φb | ||
| a Absorption and emission spectra were recorded in CH3CN at 298 K and in frozen CH3OH–C2H5OH at 77 K.b Relative luminescence quantum yield (Φ) are calculated by integrating the emission profile using [Ru(bpy)3]2+ as the standard.c kr = Φem/τ.d knr = 1/τ − kr.e ken = 1/τ − 1/τ0 (τ0 is lifetime of 1). | |||||||||
| 1 | 286 (14.8) | 606 | 159 | 0.290 | 18.23 | 0.44 | — | 590 | 0.32 |
| 333 (5.78) | |||||||||
| 429 (2.16) | |||||||||
| 458 (2.52) | |||||||||
| 3 | 286 (20.72) | 599 | 9.70 | 0.111 | 114 | 9.15 | 2.63 × 106 | 586 | 0.12 |
| 334 (9.38) | 112 | 9.91 | 0.79 | 638 | |||||
| 356 (8.12) | |||||||||
| 432 (4.58) | |||||||||
| 466 (5.62) | |||||||||
| 496 (5.32) | |||||||||
The emission band of 3 at 77 K in frozen CH3OH–C2H5OH (1:4, v/v) (Fig. 1b) is blue-shifted with noticeable increase in the emission intensity and quantum yield, characteristic of typical MLCT emitters.18 The emission profile and emission maxima change with change in the excitation wavelength at 77 K. Upon excitation at 456 nm it shows emission maxima at 638 and 586 nm revealing the presence of two emitting states. A fast energy transfer between these two states could be a reason for the decrease of luminescence lifetime and quantum yield of 3 at room temperature. Upon excitation at 496 nm the low intense band at 586 nm is suppressed with a concomitant increase in the intensity of the emission band at 638 nm. By comparing the shape of the emission bands of 3 in fluid solution at 278 K and in frozen glass it is inferred that the emission from the [(bpy)2Ru(ttpy-Izphen)] component is predominant while the emission from the [Ru(ttpy-Izphen)2] site is buried underneath the broad emission of the former at 278 K.
Spectrophotometric and spectrofluorometric titrations of the complex 3 are studied in acetonitrile–water (3:2, v/v) over the pH range 2–12 at 278 K. Concentration is kept uniform throughout the titrations by making 5 × 10−6 M solutions of the complex in Robinson–Britton buffer–acetonitrile mixture. The complex undergoes protonation at low pH followed by deprotonation on the imidazole nitrogens with increasing pH. The spectral changes that occur with change in pH and deprotonation scheme are given in the ESI.† The first deprotonation occurs when the pH is increased from 2 to 4. As a consequence, the intensity of the band at 496 nm and the trough at 400 nm decrease, while the intensity of the bands at 284 and 334 nm increase with two isobestic points at 381 and 507 nm. The second deprotonation process is observed in the pH range 8–10. The absorbance at 496 and the trough at 400 nm increase with a small red shift in the absorption maxima. The intensity of the absorption bands at 432, 466, and 334 nm decrease with the appearance of two isobestic points at 371 and 450 nm. The ground state pKa1 and pKa2 values are 2.91 ± 0.06 and 9.42 ± 0.08, respectively. The emission spectral profile is strongly dependent on the changes in pH. The complex shows luminescence switching with variation in pH from acidic, neutral, to basic with an emission enhancement factor of 2.54 and emission quenching factor of 1.82 (Fig. 2). When the pH is raised from 2 to 7 the emission intensity increases, while further increase in pH results in a decrease of the emission intensity with a significant red shift in the emission maxima. The excited state pKa1* and pKa2* values in the pH range 2–4 and 8–10 are 3.12 ± 0.09 and 8.52 ± 0.05, respectively. For the first deprotonation step, the ground state pKa1 value is lower than the corresponding excited state pKa1* value. This observation gives us an insight into the nature of the MLCT state. In acidic solution, all nitrogen atoms of the imidazole ring are protonated and the π*-energy level of the bridging ligand ttpy-Izphen is lower than that of the auxiliary bpy ligands and the excited electron goes to the ttpy-Izphen ligand from the Ru(II) center. This makes the N–H fragment of imidazole ring more basic in the excited state and hence the pKa1* value is greater than the ground state pKa1 value.19 In the second deprotonation step, the ground state pKa2 is higher when compared to the excited state pKa2* value revealing that the excited state is more acidic than the ground state. The excited electron is localised on the ancillary bpy ligands thereby making the N–H proton of imidazole ring more acidic in the excited state.
 | ||
| Fig. 2 Emission spectra of 3 (5 μM) in acetonitrile/Britton–Robinson buffer (3:2, v/v) (λex = 455 nm): (a) pH 2.00–7.00, (b) pH 7.00–12.00. | ||
The redox behavior of the complexes are investigated in deaerated acetonitrile solution to complement the spectroscopic data. The cyclic voltammetric and square wave voltammetric data of the complexes are presented in Table S1 in ESI.† The oxidation processes of the Ru(II) complexes 1 and 3 are ascribed to metal centered processes, whereas the reduction processes are ligand centered. The mononuclear complex 1 undergoes one reversible oxidation at E1/2 = 1.36 V (ΔEp = 82 mV). The trinuclear complex 3 exhibits a broad quasireversible oxidation at E1/2 = 1.34 V (ΔEp = 105 mV) for the [(bpy)2Ru(ttpy-Izphen)] component.
Another reversible but feeble oxidation process occurs at E1/2 = 0.86 V (ΔEp = 74 mV) due to the oxidation of the [Ru(ttpy-Izphen)2] center which is shifted to less positive potential compared to the parent complex [Ru(ttpy)2]2+ (E1/2 = 1.25 V).20 This is in agreement with the spectroscopic data and can be explained by the extensive delocalization of the ttpy-Izphen ligand. The quasireversibility of the Ru(II)/Ru(III) redox couple for the [(bpy)2Ru(ttpy-Izphen)] center could be a result of the prior oxidation of the [Ru(ttpy-Izphen)2]2+ component occurring at a lower potential (E1/2 = 0.86 V). An irreversible oxidation wave observed at E1/2 = 1.69 V is due to the ligand centered oxidation.21 The first reduction, usually expected to involve the ligand having the most stable lowest unoccupied molecular orbital (LUMO) is assigned to the reduction of the bridging ligand ttpy-Izphen. The following four reduction waves for 1 and three waves for 3 are characteristic of the ttpy and the peripheral bpy ligands but could not be resolved and assigned for individual processes due to their proximate appearance. It is inferred that reduction occurs first on the bridging ligand and then on the bpy ligand.
The heteroleptic homotrinuclear Ru(II) complex 3 of the ditopic imidazole-based tolylterpyridine bridging ligand with bpy ancillary ligands exhibits room temperature emission in fluid solution. Coordination of the vacant ttpy site of the mononuclear complex does not completely take toll of the luminescence by opening the non-radiative decay through 3MC state. This is due to the stabilisation of 3MLCT state by extended delocalization. It also exhibits pH-modulated luminescence switching behaviour. Time resolved photophysical studies are being carried out to supplement the understanding of excited state properties of the complex.
Acknowledgements
The authors wish to thank Sophisticated Analytical Instrumentation Facility, IIT Madras, for luminescence lifetime measurements and NMR studies. Financial assistance from the Department of Science and Technology, Government of India, is thankfully acknowledged.Notes and references
- K. L. Mulfort, A. Mukherjee, O. Kokhan, P. Duz and D. M. Tiede, Chem. Soc. Rev., 2013, 42, 2215–2227 RSC.
- (a) Md. K. Nazeeruddin, C. Klein, P. Liska and M. Grätzel, Coord. Chem. Rev., 2005, 249, 1460–1467 CrossRef CAS PubMed; (b) S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, J. Kübel, B. Dietzek, C. P. Berlinguette and U. S. Schubert, Inorg. Chem., 2014, 53, 1637–1645 CrossRef CAS PubMed.
- (a) N. Robertson and C. A. McGowan, Chem. Soc. Rev., 2003, 32, 96–103 RSC; (b) F. M. Raymo, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- (a) J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed; (b) J. D. Badijic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef PubMed; (c) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC; (d) P. Ceroni, A. Credi and M. Venturi, Chem. Soc. Rev., 2014, 43, 4068–4083 RSC.
- (a) G. R. Newkome, E. He and C. N. Moorefield, Chem. Rev., 1999, 99, 1689–1746 CrossRef CAS PubMed; (b) V. Balzani, P. Ceroni, A. Juris, M. Venturi, S. Puntariero, S. Campagna and S. Serroni, Coord. Chem. Rev., 2001, 219–221, 545–572 CrossRef CAS; (c) J.-L. Wang, X. Li, C. D. Shreiner, X. Lu, C. N. Moorefield, S. R. Tummalapalli, D. A. Medvetz, M. J. Panzner, F. R. Fronczek, C. Wesdemiotis and G. R. Newkome, New J. Chem., 2012, 36, 484–491 RSC.
- P. G. Hoertz and T. E. Mallouk, Inorg. Chem., 2005, 44, 6828–6840 CrossRef CAS PubMed.
- (a) H. B. Baudin, J. Davidsson, S. Serroni, A. Juris, V. Balzani, S. Campagna and L. Hammarstron, J. Phys. Chem. A, 2002, 106, 4312–4319 CrossRef CAS; (b) P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- (a) M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC; (b) D.-L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed.
- (a) E. Ruba, J. R. Hart and J. K. Barton, Inorg. Chem., 2004, 43, 4570–4578 CrossRef PubMed; (b) S. P. Foxon, C. Metcalfe, H. Adams, M. Webb and J. A. Thomas, Inorg. Chem., 2007, 46, 409–416 CrossRef CAS PubMed; (c) D. Hvasanov, A. F. Mason, D. C. Goldstein, M. Bhadbhade and P. Thordarson, Org. Biomol. Chem., 2013, 11, 4602–4612 RSC.
- (a) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003, ch. 2–6 and references therein Search PubMed; (b) O. S. Wenger, Coord. Chem. Rev., 2009, 253, 1439–1457 CrossRef CAS PubMed; (c) D. Hanss, M. E. Walther and O. S. Wenger, Coord. Chem. Rev., 2010, 254, 2584–2592 CrossRef CAS PubMed.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- (a) B. Jing, T. Wu, C. Tian, M. Zhang and T. Shen, Bull. Chem. Soc. Jpn., 2000, 73, 1749–1755 CrossRef CAS; (b) K.-Z. Wang, L.-H. Gao, G.-Y. Bai and L.-P. Jin, Inorg. Chem. Commun., 2002, 5, 841–843 CrossRef CAS; (c) C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed; (d) F. Cheng, J. Chen, F. Wang, N. Tang and L. Chen, J. Coord. Chem., 2012, 65, 205–217 CrossRef CAS; (e) N. Arockia Samy and V. Alexander, Dalton Trans., 2011, 40, 8630–8642 RSC.
- D. Maity, C. Bhaumik, S. Karmakar and S. Baitalik, Inorg. Chem., 2013, 52, 7933–7946 CrossRef CAS PubMed.
- U. S. Schubert, H. Hofmeier and G. Newkome, Modern Terpyridine Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) O. Hamelin, P. Guillo, F. Loiseau, M.-F. Boissonnet and S. Ménage, Inorg. Chem., 2011, 50, 7952–7954 CrossRef CAS PubMed; (b) Z.-B. Zheng, Z.-M. Duan, J.-M. Zhang and K.-Z. Wang, Sens. Actuators, B, 2012, 169, 312–319 CrossRef CAS PubMed.
- J.-Z. Wu, B.-H. Ye, L. Wang, L.-N. Ji, J.-Y. Zhou, R.-H. Li and Z.-Y. Zhou, J. Chem. Soc., Dalton Trans., 1997, 1395–1401 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133–142 RSC.
- H.-J. Mo, Y. Shen and B.-H. Ye, Inorg. Chem., 2012, 51, 7174–7184 CrossRef CAS PubMed.
- C. Bhaumik, D. Saha, S. Das and S. Baitalik, Inorg. Chem., 2011, 50, 12586–12600 CrossRef CAS PubMed.
- S. J. Slattery, N. Gokaldas, T. Mick and K. A. Goldsby, Inorg. Chem., 1994, 33, 3621–3624 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Relevant absorption, emission and excitation spectra, cyclic voltammograms, mass spectra, and NMR spectra of metal complexes. See DOI: 10.1039/c4ra07077a |
| This journal is © The Royal Society of Chemistry 2014 |