DOI:
10.1039/C4RA07031C
(Paper)
RSC Adv., 2014,
4, 34210-34216
Selenium substitution endows cystine with radiosensitization activity against cervical cancer cells†
Received
12th July 2014
, Accepted 24th July 2014
First published on 24th July 2014
Abstract
Radiotherapy has been the primary treatment for cancer along with chemotherapy and surgical therapy for decades. However, radiotherapy still fails to efficiently deracinate the hypoxic tumors because of their insensitivity to X-rays. In the present study, we report that selenocysteine (SeC), an analog of cystine (Cys) through selenium substitution of sulfur, could act as an effective radiosensitizer to enhance the anticancer efficacy of radiotherapy through induction of cancer cell apoptosis. By comparing the ROS generation activity of SeC and Cys, we found that selenium substitution significantly enhances the X-ray-induced ROS overproduction in human cervical cancer HeLa cells. Excess ROS could attack various components of DNA and activated downstream signaling pathways in HeLa cells. Specifically, SeC enhanced the radiation-induced phosphorylation of p53 and p38MAPK pathways, and down-regulation of phosphorylated AKT and ERK, and finally resulted in increased radiation sensitivity and inhibited tumor reproduction. Taken together, this study suggests that selenium substitution could be a novel strategy for design of cancer radiosensitizers.
Introduction
Cancer has been one of the most devastating diseases for decades, and radiotherapy is the primary treatment for cancer along with chemotherapy and surgical therapy. Theoretically speaking, radiotherapy uses high-energy irradiation to kill cancer cells by directly damaging the structure of DNA or creating free radicals including reactive oxygen species within the cells to attack various components of DNA, leading to the oxidized bases, DNA strand breaks, DNA intra-strand adducts, and DNA–protein crosslinks.1–6 X-ray or γ-ray radiation could focus the energy precisely on the position of tumors, thus minimized the toxic side effects to the surrounding normal tissues. Even so, radiotherapy still fail to efficiently deracinate the hypoxic tumors because of their insensitivity to X-ray.7,8 Therefore, radiosensitizers, which can enhance the sensitivity to radiotherapy, are necessary. Radiotherapy and chemotherapy are two important therapeutic modalities, only combined with these two modalities can achieve the optimum and synergetic treatment.9,10 Recently, cisplatin [Pt(NH3)2Cl2], a most widely used anti-cancer drug, have been used as a radiosensitizer in clinical use.11–13 While, the clinical drawbacks of cisplatin are apparent, including the limited applicability, the acquired resistance, the resurgence after chemotherapy, and the serious side effects, such as neurotoxicity and nephrotoxicity.14–16 In order to overcome the limitations of cisplatin, extensive investigations may be conducted to use efficient and low toxic agents from food as radiosensitizers. Recently, Lo et al. reported that, branched α-(1,4)-glucans from Lentinula edodes (L10) could enhance cytotoxic effect on human lung adenocarcinoma through the toll-like receptor 4 mediated induction of THP-1 differentiation/activation in combination with radiation.17 Chae et al. reported that, compound K, a metabolite of ginseng saponin, could suppress human lung cancer cell growth in vitro in vivo in combination with γ-ray radiation.18 Furthermore, Mallikarjunan et al. also demonstrated the influence of γ-radiation on the structure and function of soybean trypsin inhibitor.19 Taken together, these results support the application potential of food constituents as radiosensitizers in radiotherapy.
Selenium (Se) is one of the essential trace elements for humans and animals with important physiological functions and extensive pharmacological actions. It was found that there are various organic forms of Se in food, such as selenomethionine, Se-methyl-selenocysteine, selenocysteine and its disulfide dimer selenocysteine.20 Se is also the important component of Se-dependent enzymes, such as peroxidase, iodothyronine deiodinase, and thioredoxin reductase.21 Many studies have demonstrated that the moderate supplementation of Se could prevent the incidence of cancer.22–25 In recent years, substantial amount of evidences showed the potent antiproliferative activity of Se.26–29 For instance, Schüller et al. suggested a radiosensitizing effect of selenite in glioma cells at low concentrations.30 Hu and co-workers reported that selenocompounds could sensitize hormone refractory prostate cancer to paclitaxe in vitro and in vivo, showing that Se compounds and taxane may be achieve synergetic treatment.31 Wu et al. synthesized surface-capping selenium nanoparticles by polysaccharide–protein complexes from polyporus rhinoceros and found that they can induce the apoptosis and cell cycle arrest in A549 human lung adenocarcinoma cells.32 Recently, we also found that, modification of Se nanoparticles by amino acids could enhance its anticancer efficacy,33 which suggest the synergistic anticancer action between amino acids and Se.
Since sulfur and Se are in the same group (VIA) of periodic table of elements, it is feasible to substitute sulfur atom in active compounds with Se. Recent studies have reported that substituting sulfur with Se in established chemopreventive agents may lead to more effective analogs.34–36 D-501036, a novel selenophene-based triheterocycle, has been identified as a promising anticancer compound with potential for therapy of human cancers.35 Moreover, Desai and co-workers verified that substitution of sulfur with Se increased the compound's potency by several folds in different cancer cell lines by inhibition of iNOS/AKT.37 Selenocysteine (SeC), an analog of cystine (Cys) through Se substitution of sulfur, displayed selective anticancer effects on a number of human cancer cell lines. For example, Chen et al. reported that SeC induced apoptosis of A375 human melanoma cells by activating reactive oxygen species (ROS)-mediated mitochondrial pathway and p53 phosphorylation.29 They also reported that SeC triggered S-phase arrest and apoptosis in human breast adenocarcinoma MCF-7 cells by modulating ERK and AKT phosphorylation.38 These results imply the potential of SeC as a cancer chemopreventive and chemotherapeutic agent. However, little information about the radiosensitizing effects of SeC is available. Therefore, in this study, we showed that, SeC sensitized cervical cancer HeLa cells to X-ray and enhanced the cell growth inhibition through induction of caspase-mediated apoptosis. The studies on the signaling pathways demonstrate that, intracellular SeC synergize with X-ray to trigger ROS overproduction. Excessive ROS induces DNA damage and activates downstream signaling pathways, finally results in increased radiation sensitivity and thus inhibits the tumor reproduction. Taken together, this study suggests Se substitution could be a novel strategy for design of cancer radiosensitizers, and SeC could be further developed as an effective and low toxic cervical cancer radiosensitizer.
Experimental sections
Materials and chemicals
Thiazolyl blue tetrazolium bromide (MTT), propidium iodide, 4′,6-diamidino-2-phenyindole (DAPI) and bicinchoninic acid (BCA) kit were purchased from Sigma. Substrates for caspase-3 (Ac-DEVD-AMC), caspase-8 (Ac-IETD-AFC) and caspase-9 (Ac-LEHD-AFC) were purchased from Calbiochem. Terminal transferase dUTP nick end labeling (TUNEL) assay kit was purchased from Roche Molecular Biochemicals. X-ray radiation was carried on Elekta Precise linear accelerator in Wu Jing Zong Dui Hospital of Guangdong Province, China.
Cell lines and cell culture
Human cervical cancer cell lines, including HeLa, Caski and Siha cells, were purchased from American Type Culture Collection (ATCC, Manassas, VA). This cell line was maintained in DMEM media supplemented with fetal bovine serum (10%), penicillin (100 units per mL) and streptomycin (50 units per mL) at 37 °C in CO2 incubator (95% relative humidity, 5% CO2).
Clonogenic assays
HeLa cells were seeded into 6-well plates at 1000 cells per mL (2 mL) and allowed to attach for 24 h. After 2 h incubation with SeC or Cys at different concentrations, the cells were then exposed to X-ray at 2 Gy and cultured up to 8 days. After that the cells were stained with 6.0% glutaraldehyde (v/v), 0.5% crystal violet (w/v) in water and photographed colonies of greater than 50 cells were counted. The survival fraction was calculated using the following formula: ((number of colonies formed after radiation)/(number of cells seeded × plating efficiency)), where plating efficiency is the ratio of seeded cells that gave rise to clones under no radiation conditions.
MTT assay
The effects of SeC, Cys and X-ray on the cell viability was determined by MTT assay as previously described.28
Flow cytometric analysis
The effects of SeC enhances radiation-induced apoptosis was analyzed by flow cytometry as previously described.39
TUNEL and DAPI co-staining assay
HeLa cells treated with or without SeC and X-ray for 24 h were fixed with 3.7% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100 in PBS. The cells were then incubated with 100 μL per well TUNEL for 1 h and 1 μg mL−1 of DAPI for 15 min at 37 °C, respectively. DNA fragmentation and nuclear condensation was examined under a fluorescence microscope (Nikon Eclipse 80i).40
Determination of caspase activity
Caspase activity was measured as previously described.41 Briefly, after preparation for the whole cell lysis, the cell lysates were placed in 96-well plates and then specific caspase-8, -9 substrate (Ac-LEHD-AMC) was added. Plates were incubated at 37 °C for 2 h and caspase activity was determined by fluorescence intensity with the excitation and emission wavelengths set at 380 and 460 nm, respectively.
Measurement of intracellular reactive oxygen species generation
The effects of SeC and radiation on intracellular ROS generation in HeLa cells were monitored by DCF-DA assay.27 Briefly, HeLa cells with a density of 5 × 104 cells per well were seeded in 96-well tissue culture plates for 24 h. Cells were then treated with SeC at different concentrations at 37 °C for 2 h, 10 μM DCF-DA for 30 min, and then co-treated with or without X-ray (2 Gy). The intracellular ROS level was examined under a microplate reader (SpectraMax M5, MD, USA) with the excitation and emission wavelengths at 479 and 599 nm, respectively.
Western blot analysis
Total cellular proteins were extracted by incubating cells in lysis buffer obtained from Cell Signaling Technology and protein concentrations were determined by BCA assay. The effects of SeC enhances radiation on the expression levels of proteins associated with different signaling pathways were examined by western blot analysis.42
Statistics analysis
All experiments were carried out at least in triplicate, and the data are expressed as mean ± standard deviation. Differences between the control and the experimental groups were analyzed by two-tailed Student's t test. One-way analysis of variance (ANOVA) was used in multiple group comparisons. Statistical analysis was performed using SPSS statistical program version 13 (SPSS Inc., Chicago, IL). Significant difference between treatment and control groups is indicated at *P < 0.05, **P < 0.01 level.
Results and discussion
Se substitution endows cystine radiosensitization activity
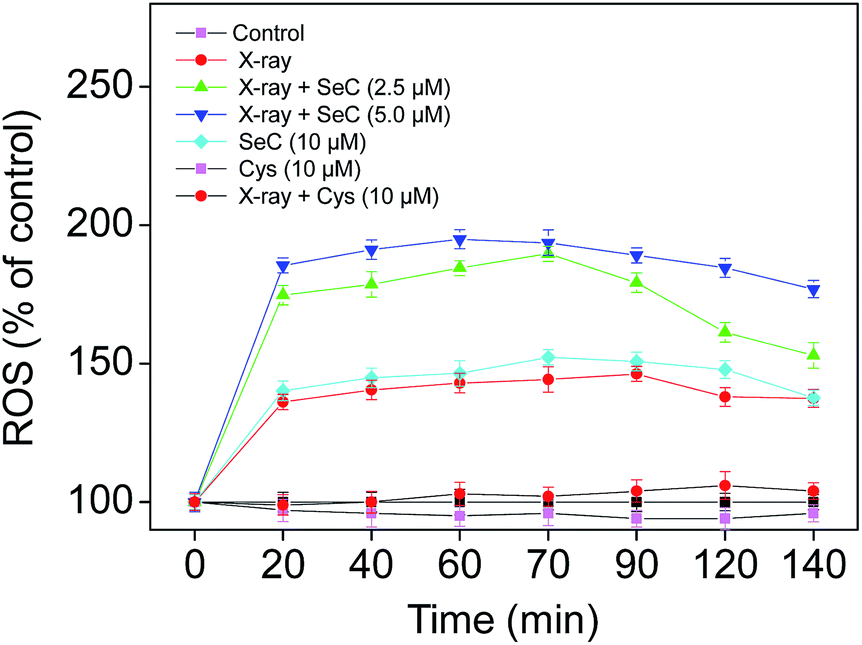
Cervical cancer has becoming a serious problem and leading cause of cancer-related death in women. Most traditional treatment to cervical cancer patients are standard radiotherapy and chemotherapy. However, radiotherapy may still fail to efficiently deracinate the hypoxic tumors because of their insensitivity to X-ray.7,8 In this study, we have used MTT assay to evaluate the cell viability of HeLa cells exposed to different doses of X-ray with or without SeC. As shown in Fig. S1A,† the X-ray radiation was not cytotoxic toward HeLa cells. The cell viability of HeLa cells were maintained at about 95.8–92.1% after exposure to 1, 2 and 4 Gy of X-ray. The results of flow cytometric also demonstrated that the X-ray radiation showed no effects on the cycle arrest distribution of HeLa cells (Fig. S1B†). In order to enhance the sensitivity of HeLa cells to radiation, an effective radiosensitizer to enhance the effects of radiotherapy is necessary. Fortunately, in this study, we found that, SeC effectively enhanced the sensitivity of human cervical cancer cell lines, including HeLa, Caski and Siha cells, to X-ray radiation. The combined treatment resulted in significant reduction of cell viability, by comparing with X-ray or SeC alone (Fig. 1C and D and S3 and 4†). In contrast, Cys under the same concentrations showed no effects on the anticancer action of X-ray. The significant difference demonstrate the obvious effects of Se substitution on the anticancer action of Cys. Furthermore, we evaluated the effects of Se substitution on the radiotherapeutic activity of Cys by clonogenic assay. As shown in Fig. 1A, the images of cell colony formation in 6-well plates indicated that SeC enhanced the radiation-induced inhibition of colony formation. The survival fraction of HeLa cells treated by X-ray (2 Gy) and SeC (20 μM) alone were 93.4% and 41.7%, respectively. While after co-treated with X-ray and SeC at 5, 10 and 20 μM, the survival fraction was continuously declined to 5.9% (Fig. S2†). However, Cys couldn't inhibit the cell colony formation, even under co-treatment with X-ray (Fig. 1B). The survival fraction of HeLa cells after incubation with Cys and X-ray both exhibited no changes by comparing with control (Fig. S2†). Moreover, MTT assay was used to further confirm the enhancement effects of Se substitution. As shown in Fig. 1C, the significant difference of the cell viability among the combined treatment and SeC or X-ray alone was observed. The cell viability treated by X-ray (2 Gy) and SeC (20 μM) alone were 90.26% and 35.40%, respectively. While after co-treated with X-ray and SeC at 5, 10 and 20 μM, the cell viability has significantly declined to 10.62%, 2.65% and 0.88%, respectively. The morphological changes (Fig. S3†) of HeLa cells also demonstrated that SeC can sensitizes cancer cells to X-ray. In contrast, as shown in Fig. 1D, treatments of the cells with Cys or even in combination with X-ray showed no significant growth inhibitory effects. Furthermore, significant enhancement of X-ray-induced cell growth inhibition was also observed in Caski and Siha cells exposed to SeC (Fig. S4†). Taken together, these results suggest that Se substitution endows cystine radiosensitization activity against cervical cancer cells.
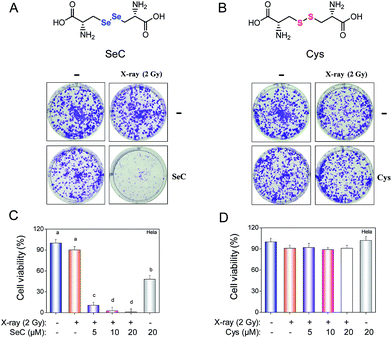 |
| | Fig. 1 Se substitution endows Cys radiosensitization activity. (A) Clonogenic assay performed in 6 well plates cultured up to 8 days after treated with 20 μM SeC or X-ray. (B) Clonogenic assay performed in 6 well plates cultured up to 8 days after treated with 20 μM Cys or X-ray. (C) Viability of HeLa cells exposed to the combined treatment and SeC or X-ray alone, and then cultured for 24 h. (D) Viability of treated HeLa cells under the same conditions. Cell viability was determined by MTT assay. | |
Se substitution enhances radiation-induced ROS generation
Theoretically, radiotherapy uses ionizing radiation to kill cancer cells by directly damaging DNA structure or by creating free radicals within the cells to damage the DNA.4–6 ROS was mainly produced from mitochondrial respiratory chain in cells, especially superoxide and hydrogen peroxide.43 Many studies have demonstrated that Se could stimulate cancer cells to produce excess ROS, which could attack various components of DNA, cause DNA damage, thus induce the apoptosis in cancer cells.27,29,44,45 For instance, Stewart et al. have compared the catalytic and superoxide-producing properties of three selenocompounds, and found that sodium selenite and selenocystamine generated superoxide and induced apoptosis of BALB/c MK2 cells. In contrast, selenomethionine didn't generate superoxide, and thus was not cytotoxic to the cells.46 Furthermore, our previous studies have showed that, SeC induced apoptosis in A375 human melanoma cells and MCF-7 human breast adenocarcinoma cells by activating ROS-mediated mitochondrial pathway.27–29,38 Therefore, in this study, we tried to examine the roles of ROS in the radiosensitization action of SeC. Briefly, we have detected the intracellular ROS generation in HeLa cells treated with SeC or Cys under X-ray irradiation by DCF-DA assay. As shown in Fig. 2, treatment of HeLa cells with X-ray (2 Gy) alone displayed increase in DCF fluorescence intensity in a time-dependent manner. However, the ROS level displayed a much higher increase after combined treatment with SeC and X-ray. In contrast, treatment of the cells with Cys alone slightly inhibited the intracellular ROS generation, while the combined treatment with Cys and X-ray only slightly increased the ROS level. This significant difference between SeC and Cys may be caused by the Auger electron effect of Se atom under X-ray radiation. Taken together, these results suggest that, Se substitution of sulfur in Cys may sensitize the cancer cells to X-ray through enhanced induction of ROS generation.
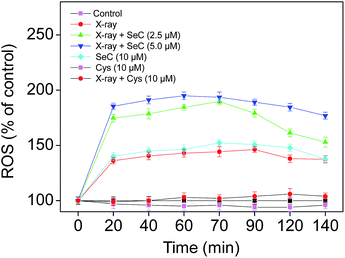 |
| | Fig. 2 Se substitution enhances radiation-induced ROS generation. Cells were treated with SeC or Cys for 2 h, and then co-treated with X-ray (2 Gy). After different period of time, the intracellular ROS generation was determined by DCFH-DA fluorescence assay. | |
SeC enhances radiation-induced cell apoptosis through caspase activation
Apoptosis, cell cycle arrest or combination of these two modes are possible causes of inhibition of cell proliferation.47 To further evaluate mechanism of radiosensitization by SeC, flow cytometric analysis was used to examine the DNA distribution after treated with or without X-ray and SeC. As shown in Fig. 3A, the representative DNA histograms clearly showed that the significant difference of exposure of HeLa cells among the combined treatment and SeC or X-ray alone. For the sub-G1 phase, there was no big difference between the single X-ray (2 Gy), SeC (5, 10 μM) and the control. However, after co-treated with X-ray and SeC (5, 10 μM), the sub-G1 peak increased from 2.7% (control) to 53.6% and 53.8%, respectively. The induction of apoptosis was further confirmed by TUNEL-DAPI co-staining assay, which can detect early stage of DNA fragmentation and nuclear condensation in apoptotic cells. As shown in Fig. 3B, after co-treated with X-ray and SeC (5 μM), HeLa cells exhibited typical apoptotic features including nuclear condensation, chromatin condensation and formation of apoptotic bodies. While HeLa cells treated with X-ray alone didn't exhibit the obvious apoptotic features, and cells treated with SeC (5 μM) alone also exhibited weak apoptotic features. Taken together these results indicate that SeC could enhance radiation-induced apoptosis in human cervical cancer cells.
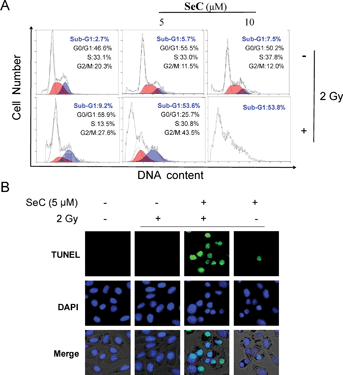 |
| | Fig. 3 SeC enhances radiation-induced apoptosis in HeLa cells. (A) Flow cytometric analysis of HeLa cells treated by SeC and X-ray (2 Gy). HeLa cells treated with or without SeC and X-ray for 24 h were collected and stained with PI. Cellular DNA histograms were analyzed by flow cytometer. (B) DNA fragmentation and nuclear condensation induced by radiation and SeC as determined by TUNEL-DAPI assay (magnification, 400×). | |
To delineate the molecular events of radiosensitization by SeC, we examined the requirement of caspase family proteins for the apoptotic program. Generally, in mammalian cells, initiator caspases, caspase-8 and caspase-9 would activate caspase-3 by proteolytic cleavage, which then cleaves vital cellular proteins or other caspases. In this study, from the results of western blot, we found that HeLa cells co-treated with X-ray and SeC caused activation of caspase-3, caspase-8 and caspase-9 (Fig. 4A). Poly(ADP-ribose) polymerase (PARP) is a DNA repair enzyme. In apoptosis pathways, PARP is the downstream of caspase family proteins. Caspase-3 activation subsequently induced the cleavage of PARP, which serves as a biochemical marker of the cell apoptosis. As shown in Fig. 4A, we also detected the proteolytic cleavage of PARP in HeLa cells after treated with X-ray and SeC. Caspase-3 has been regarded as the central regulator of apoptosis, while caspase-8 and -9 acted as initiators of extrinsic and intrinsic signaling pathways. To further examine the importance of caspase-8 and caspase-9, we examined the activities of the aforementioned caspases. As shown in Fig. 4B, exposure to X-ray (2 Gy) and SeC (2 μM) significantly increased the activities of caspase-8 and -9, whereas no significant changes was observed in HeLa cells single treated with X-ray and SeC. It is thus speculated that the caspase-mediated apoptosis is the main signaling pathway triggered by X-ray and SeC.
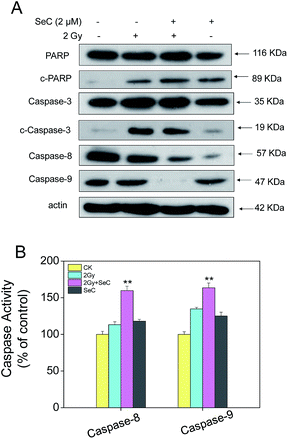 |
| | Fig. 4 Activation of caspases by SeC and X-ray. (A) Western blot analysis of expression levels of the caspase family members after treated by SeC and X-ray for 24 h. (B) Quantitative analysis of caspase activation triggered by radiation and SeC. Cells were treated with SeC and X-ray for 24 h. Significant difference between treatment and control groups is indicated at *P < 0.05, **P < 0.01 level. | |
SeC enhances radiation-induced ROS-mediated signaling
Studies have showed that excess intracellular ROS could activate the MAPK and AKT pathway in cancer cells.48 Therefore we next examined the MAPK and AKT expression and phosphorylation in HeLa cells after treated with or without X-ray and SeC by western blot analysis. It was found that X-ray and SeC treatment induced differential phosphorylation of p38, JNK, ERK, and AKT in HeLa cells (Fig. 5). The figure showed that phosphorylation of pro-apoptotic kinases p38 and JNK activation displayed up-regulation co-treated with SeC and X-ray compare to that single treated with X-ray or SeC. In addition, phosphorylation of anti-apoptotic kinases ERK was activated by co-treated with SeC and X-ray and displayed down-regulation. AKT pathway plays a role in cell growth inhibition, the expression of phosphorylated AKT displayed a trend of down-regulation in HeLa cells after co-treated with SeC and X-ray. These results suggested that SeC enhances the anticancer efficacy of X-ray through activation of MAPKs and AKT pathways.
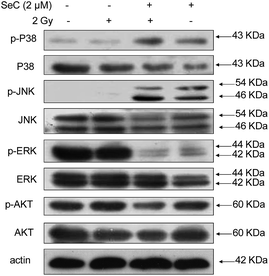 |
| | Fig. 5 SeC enhances radiation-induced ROS-mediated signaling. Effects of SeC and radiation on the phosphorylation status and expression levels of MAPKs and AKT pathways. | |
SeC synergizes with X-ray to induce DNA damage-mediated signaling
The previous results showed that X-ray and SeC treatment induced ROS-mediated DNA damage in HeLa cells, suggesting the possible involvement of p53 pathway. To examine whether p53 is activated by SeC and radiation, we determined the p53 expression and phosphorylation in HeLa cells after treated with or without SeC and X-ray. As shown in Fig. 6A, both SeC and X-ray can activate the phosphorylated p53 at Ser 15 in HeLa cells. Protein levels of a DNA damage marker Ser 139–Histone H2A.X were up-regulated much more co-treated with SeC and X-ray than that single treated with them. Furthermore, from the expression levels of p-ATM and p-ATR, the radiation-induced DNA damage was further demonstrated. However, after co-treated with SeC, the expression levels significantly up-regulated. These results further indicate that SeC enhances radiation-induced DNA damage and the activation of p53 pathway is required for SeC and radiation-induced apoptosis. VEGFR2 is overexpressed in cervical cancer cells, and it's expression often act as the reference index for migration, invasion and prognosis of cervical cancer.49 Then, we examined the expression levels of VEGFR2 and found that X-ray and SeC treatment induced it's down-regulation (Fig. 6B). However, most radiation-induced DNA damage is sublethal, and the cells are able to repair themselves in lower doses.50 The X-ray repair cross complementing protein 1 (XRCC1),51,52 a DNA repair protein, was evaluated after treated with X-ray and SeC by western blot analysis. As shown in Fig. 6B, the expression levels of XRCC1 significantly up-regulated after treated with single X-ray (2 Gy). However, SeC can reduce X-ray-induced up-regulation of XRCC1, which may be beneficial to prevent the re-occurrence of cervical cancer. Taken together these results indicated that activation of p53 pathway is required for SeC and radiation-induced apoptosis.
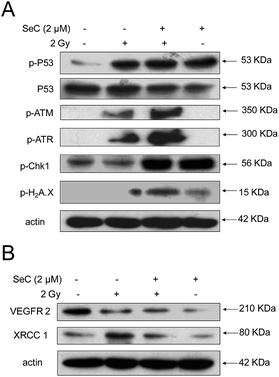 |
| | Fig. 6 SeC synergizes with X-ray to induce DNA damage-mediated signaling. (A) SeC enhances X-ray-induced DNA damage-mediated p53 phosphorylation. (B) SeC down-regulates the expression of VEGFR2 and XRCC1. | |
Conclusions
In conclusion, this study identifies SeC, an analog of Cys through Se substitution of sulfur, could act as an effective radiosensitizer to enhance the anticancer efficacy of X-ray. SeC sensitized human cervical cancer HeLa cells to X-ray and enhanced the cell growth inhibition through induction of caspase-mediated apoptosis. We also proposed the signaling pathways accounting for the synergistic effects of X-ray radiation and SeC (Fig. 7). Firstly, intracellular SeC synergize with X-ray to trigger ROS overproduction. Excessive ROS induces DNA damage and activate downstream signaling pathways, including the phosphorylation of p53 and p38MAPK and down-regulation of phosphorylated AKT and ERK, finally results in increased radiation sensitivity and thus inhibits the tumor reproduction. Taken together, this study suggests Se substitution could be a novel strategy for design of cancer radiosensitizers, and SeC could be further developed as an effective and low toxic cervical cancer radiosensitizer.
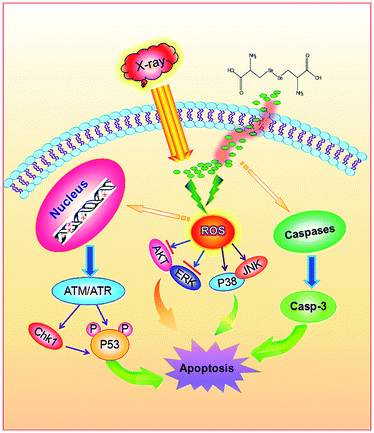 |
| | Fig. 7 Proposed apoptotic signaling pathways induced by radiation and SeC. | |
Acknowledgements
This work was supported by National High Technology Research and Development Program of China (863 Program, SS2014AA020538), Science Foundation for Distinguished Young Scholars of Guangdong Province, Natural Science Foundation of China and Research Fund for the Doctoral Program of Higher Education of China.
Notes and references
- C. Beskow, J. Skikuniene, A. Holgersson, B. Nilsson, R. Lewensohn, L. Kanter and K. Viktorsson, Br. J. Cancer, 2009, 101, 816–821 CrossRef CAS PubMed.
- G. Afshar, N. Jelluma, X. Yang, D. Basila, N. D. Arvold, A. Karlsson, G. L. Yount, T. B. Dansen, E. Koller and D. A. Haas-Kogan, Cancer Res., 2006, 66, 4223–4232 CrossRef CAS PubMed.
- W. Fan, B. Shen, W. Bu, F. Chen, K. Zhao, S. Zhang, L. Zhou, W. Peng, Q. Xiao, H. Xing, J. Liu, D. Ni, Q. He and J. Shi, J. Am. Chem. Soc., 2013, 135, 6494–6503 CrossRef CAS PubMed.
- T. Takeshima, K. Chamoto, D. Wakita, T. Ohkuri, Y. Togashi, H. Shirato, H. Kitamura and T. Nishimura, Cancer Res., 2010, 70, 2697–2706 CrossRef CAS PubMed.
- E. I. Azzam, S. M. de Toledo and J. B. Little, Curr. Cancer Drug Targets, 2004, 4, 53–64 CrossRef CAS.
- C. J. Lord and A. Ashworth, Nature, 2012, 481, 287–294 CrossRef CAS PubMed.
- J. F. Hainfeld, F. A. Dilmanian, D. N. Slatkin and H. M. Smilowitz, J. Pharm. Pharmacol., 2008, 60, 977–985 CrossRef CAS PubMed.
- T. Nakae, Y. Uto, M. Tanaka, H. Shibata, E. Nakata, M. Tominaga, H. Maezawa, T. Hashimoto, K. L. Kirk, H. Nagasawa and H. Hori, Bioorg. Med. Chem., 2008, 16, 675–682 CrossRef CAS PubMed.
- H. Hoshikawa, K. Indo, T. Mori and N. Mori, Cancer Lett., 2011, 306, 60–66 CrossRef CAS PubMed.
- C. Farrell and E. Lennan, NursePrescribing, 2013, 11, 353–358 Search PubMed.
- A. A. Forastiere, H. Goepfert, M. Maor, T. F. Pajak, R. Weber, W. Morrison, B. Glisson, A. Trotti, J. A. Ridge, C. Chao, G. Peters, D. J. Lee, A. Leaf, J. Ensley and J. Cooper, N. Engl. J. Med., 2003, 349, 2091–2098 CrossRef CAS PubMed.
- N. E. Marshall, K. V. Ballman, J. C. Michalak, P. J. Schomberg, G. V. Burton, H. M. Sandler, T. L. Cascino, K. A. Jaeckle and J. C. Buckner, J. Neuro-Oncol., 2006, 77, 315–320 CrossRef CAS PubMed.
- S. Cakir and I. Egehan, Lung Cancer, 2004, 43, 309–316 CrossRef PubMed.
- M. Markman, Expert Opin. Drug Saf., 2003, 2, 597–607 CrossRef CAS.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- T. C.-T. Lo, F.-M. Hsu, C. A. Chang and J. C.-H. Cheng, J. Am. Chem. Soc., 2011, 59, 11997–12005 CAS.
- S. Chae, K. A. Kang, W. Y. Chang, M. J. Kim, S. J. Lee, Y. S. Lee, H. S. Kim, D. H. Kim and J. W. Hyun, J. Agric. Food Chem., 2009, 57, 5777–5782 CrossRef CAS PubMed.
- N. Mallikarjunan, S. Marathe, R. Deshpande, S. N. Jamdar and A. Sharma, J. Agric. Food Chem., 2012, 60, 12036–12043 CrossRef CAS PubMed.
- M. Bjornstedt, S. Kumar, L. Bjorkhem, G. Spyrou and A. Holmgren, Biomed. Environ. Sci., 1997, 10, 271–279 CAS.
- H. Song, I. Hur, H. J. Park, J. Nam, G. B. Park, K. H. Kong, Y. M. Hwang, Y. S. Kim, D. H. Cho, W. J. Lee and D. Y. Hur, Immune. Netw., 2009, 9, 236–242 CrossRef PubMed.
- J. W. Card and B. A. Magnuson, Int. J. Toxicol., 2010, 29, 402–410 CrossRef CAS PubMed.
- J. Zhang, H. Wang, X. Yan and L. Zhang, Life Sci., 2005, 76, 1099–1109 CrossRef CAS PubMed.
- C. H. Takimoto, M. A. Graham, G. Lockwood, C. M. Ng, A. Goetz, D. Greenslade, S. C. Remick, S. Sharma, S. Mani, R. K. Ramanathan, T. W. Synold, J. H. Doroshow, A. Hamilton, D. L. Mulkerin, P. Ivy, M. J. Egorin and J. L. Grem, Clin. Cancer Res., 2007, 13, 4832–4839 CrossRef CAS PubMed.
- J. A. Martenson, S. R. Lipsitz, H. Wagner Jr, E. H. Kaplan, L. A. Otteman, L. M. Schuchter, E. G. Mansour, M. S. Talamonti and A. B. Benson 3rd, Int. J. Radiat. Oncol., Biol., Phys., 1996, 35, 745–749 CrossRef CAS.
- O. Micke, L. Schomburg, J. Buentzel, K. Kisters and R. Muecke, Molecules, 2009, 14, 3975–3988 CrossRef CAS PubMed.
- T. Chen and Y. S. Wong, Int. J. Biochem. Cell Biol., 2009, 41, 666–676 CrossRef CAS PubMed.
- T. Chen and Y. S. Wong, Biomed. Pharmacother., 2009, 63, 105–113 CrossRef CAS PubMed.
- T. Chen and Y. S. Wong, Cell. Mol. Life Sci., 2008, 65, 2763–2775 CrossRef CAS PubMed.
- P. Schüller, S. Püttmann, O. Micke, V. Senner, U. Schäfer and N. Willich, Trace Elem. Electrolytes, 2005, 22, 201–206 CrossRef.
- H. Hu, G. X. Li, L. Wang, J. Watts, G. F. Combs Jr and J. Lu, Clin. Cancer Res., 2008, 14, 1150–1158 CrossRef CAS PubMed.
- H. Wu, H. Zhu, X. Li, Z. Liu, W. Zheng, T. Chen, B. Yu and K. H. Wong, J. Agric. Food Chem., 2013, 61, 9859–9866 CrossRef CAS PubMed.
- Y. Feng, J. Su, Z. Zhao, W. Zheng, H. Wu, Y. Zhang and T. Chen, Dalton Trans., 2014, 43, 1854–1861 RSC.
- J. B. Hudson, L. Harris, R. J. Marles and J. T. Arnason, Photochem. Photobiol., 1993, 58, 246–250 CrossRef CAS PubMed.
- S. H. Juang, C. C. Lung, P. C. Hsu, K. S. Hsu, Y. C. Li, P. C. Hong, H. S. Shiah, C. C. Kuo, C. W. Huang, Y. C. Wang, L. Huang, T. S. Chen, S. F. Chen, K. C. Fu, C. L. Hsu, M. J. Lin, C. J. Chang, C. L. Ashendel, T. C. Chan, K. M. Chou and J. Y. Chang, Mol. Cancer Ther., 2007, 6, 193–202 CrossRef CAS PubMed.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailander, Macromol. Biosci., 2008, 8, 1135–1143 CrossRef CAS PubMed.
- D. Desai, S. V. Madhunapantula, K. Gowdahalli, A. Sharma, R. Chandagaludoreswamy, K. El-Bayoumy, G. P. Robertson and S. Amin, Bioorg. Med. Chem. Lett., 2010, 20, 2038–2043 CrossRef CAS PubMed.
- T. Chen and Y. S. Wong, J. Agric. Food Chem., 2008, 56, 10574–10581 CrossRef CAS PubMed.
- L. M. Wilhelmsson, F. Westerlund, P. Lincoln and B. Norden, J. Am. Chem. Soc., 2002, 124, 12092–12093 CrossRef CAS PubMed.
- T. Chen, Y. Liu, W. J. Zheng, J. Liu and Y. S. Wong, Inorg. Chem., 2010, 49, 6366–6368 CrossRef CAS PubMed.
- W. Liu, X. Li, Y. S. Wong, W. Zheng, Y. Zhang, W. Cao and T. Chen, ACS Nano, 2012, 6, 6578–6591 CrossRef CAS PubMed.
- S. Liu, W. Cao, L. Yu, W. Zheng, L. Li, C. Fan and T. Chen, Dalton Trans., 2013, 42, 5932–5940 RSC.
- M. Zhang, P. C. Cheung and L. Zhang, J. Agric. Food Chem., 2001, 49, 5059–5062 CrossRef CAS PubMed.
- C. Fan, J. Chen, Y. Wang, Y. S. Wong, Y. Zhang, W. Zheng, W. Cao and T. Chen, Free Radical Biol. Med., 2013, 65, 305–316 CrossRef CAS PubMed.
- C. Fan, W. Zheng, X. Fu, X. Li, Y. S. Wong and T. Chen, Oncotarget, 2014, 5, 2853–2863 Search PubMed.
- M. S. Stewart, J. E. Spallholz, K. H. Neldner and B. C. Pence, Free Radical Biol. Med., 1999, 26, 42–48 CrossRef CAS.
- C. Fan, J. Jiang, X. Yin, K.-H. Wong, W. Zheng and T. Chen, Food Chem., 2012, vol. 134, 253–261 CrossRef PubMed.
- L. Li, Y. S. Wong, T. Chen, C. Fan and W. Zheng, Dalton Trans., 2012, 41, 1138–1141 RSC.
- V. M. Nagy, R. Buiga, I. Brie, N. Todor, O. Tudoran, C. Ordeanu, P. Virag, O. Tarta, M. Rus and O. Balacescu, Rom. J. Morphol. Embryol., 2011, 52, 53–59 Search PubMed.
- G. H. Nguyen, M. M. Murph and J. Y. Chang, Cancers, 2011, 3, 1232–1252 CrossRef PubMed.
- R. Brem and J. Hall, Nucleic Acids Res., 2005, 33, 2512–2520 CrossRef CAS PubMed.
- T. Iftner, M. Elbel, B. Schopp, T. Hiller, J. I. Loizou, K. W. Caldecott and F. Stubenrauch, EMBO J., 2002, 21, 4741–4748 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4ra07031c |
| ‡ These authors contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.