
Porous metal–organic framework MIL-100(Fe) as an efficient catalyst for the selective catalytic reduction of NOx with NH3†
Peng Wanga,
Huimin Zhaoa,
Hong Sunb,
Hongtao Yua,
Shuo chena and
Xie Quan*a
aSchool of Environmental Science and Technology, Dalian University of Technology, Key Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education, China), Dalian, 116024, China. E-mail: quanxie@dlut.edu.cn; Fax: +86-411-84706263
bSchool of Environmental & Chemical Engineering, Dalian Jiaotong University, Dalian 116028, China
First published on 29th September 2014
Abstract
The development of an efficient catalyst with excellent catalytic activity and high SO2 resistance at low temperatures (<300 °C) remains a challenge for the selective catalytic reduction (SCR) reaction. In this study, we report that MIL-100(Fe) is an alternative catalyst for the SCR of NOx with NH3, and it exhibits higher NOx conversion than the conventional V2O5–WO3/TiO2 catalyst below 300 °C. In addition, the effect of H2O and SO2 on the catalytic activity is reversible, and NOx conversions are recovered after ceasing the flow of SO2 and H2O. In situ diffuse reflectance infrared Fourier transform spectroscopy results indicate that both ionic NH4+ and coordinated NH3 existed on MIL-100(Fe) and that the reaction primarily followed the Langmuir–Hinshelwood mechanism, in which NH4+ reacts with NO2 formed from NO oxidation over iron sites. Furthermore, the redox properties of the iron species (Fe3+ + e− ↔ Fe2+) could play a significant role in activating the reactants.
1. Introduction
The selective catalytic reduction (SCR) of NOx with NH3 is an efficient method for removing NOx from stationary sources.1–3 In practice, the most widely used catalyst for this process is V2O5–WO3(MoO3)/TiO2, which is effective at high operating temperatures (300–400 °C).3 Therefore, the SCR unit must be located upstream from the electrostatic precipitator. However, this placement accelerates the deactivation of the catalyst due to the high concentrations of SO2 and ash. In addition, this type of catalyst is still not satisfactory due to the volatility and toxicity of vanadium as well as the high rate of conversion of SO2 to SO3.4 Therefore, substantial research has been devoted to the development of new vanadium-free catalysts for low temperature SCR (<300 °C) such that the SCR unit can be located downstream of the electrostatic precipitator and desulfurizer.At present, the development of efficient SCR catalysts especially low temperature catalysts causes extensive attention. The transition metal oxide supported catalysts or combined catalysts as Ce-based catalyst, Cu-based catalysts, and Mn-based catalysts displayed good catalytic activity compared with the conventional V2O5–WO3/TiO2 catalysts in the SCR process.5–10 However, the catalysts exhibit poor SO2 tolerance, especially at relatively low temperature.11 Therefore, a catalyst with both high activity and high sulfur resistance must be developed.
Metal–organic frameworks (MOFs), which are known to be porous crystalline materials with a large surface area, have recently attracted increasing attention as heterogeneous catalysts due to their unique properties (i.e., high metal content and porosity).12 MOFs have an infinite network structure constructed of multitopic organic ligands and metal ions, and the pore structure can be customized using the appropriate linkers.13 The high degree of tunability along with a high surface area and pore topology indicate that MOFs with transition metal centers might serve as potential catalysts for the SCR reaction, especially in the presence of SO2, because the large surface area and porosity are beneficial for dispersing the catalytic components and reducing the accumulation of sulfates on the surface to improve catalytic activity and SO2 resistance.14 Another important feature that allows MOFs to serve as SCR catalysts is the materials' high metal content; moreover, in MOFs, all of the active sites are ultra-highly dispersed and homogeneous due to the highly crystalline nature of the materials.15–17 Active sites are freely accessible to reagent molecules. In addition, because the metal ions in MOFs are bonded to organic linkers, the combination of metal cations and organic linkers prevents SO2 poisoning of the active sites.18 Therefore, MOFs with transition metals are anticipated to be efficient and sulfur-resistant catalysts for the SCR.
Iron-based catalysts, such as Fe oxides or Fe3+ exchange zeolites, are active and environmentally friendly for the NH3-SCR due to their good activity and N2 selectivity.19–21 Therefore, MOFs constructed with iron centers could be an alternative SCR catalyst that exhibits high catalytic activity and high SO2 resistance. MIL-100(Fe) (MIL: Materials of Institute Lavoisier) is a three-dimensional iron(III) trimesate that is composed of trimers of iron octahedra sharing a common vertex μ3-O; these trimers are linked by benzene-1,3,5-tricarboxylate moieties to form hybrid supertetrahedra.22 MIL-100(Fe) contains a significant amount of FeIII CUS (coordinatively unsaturated metal sites) and FeII CUS, which are due to thermal activation in an inert gas flow, that act as Lewis acid sites.23 These sites facilitate the adsorption of reactants and promote catalysis. In addition, MIL-100(Fe) could kept the stable structure in the condition with the existence of NOx, NH3, SO2, H2O and CO2.24–27 Therefore, MIL-100(Fe) could be a stable catalyst in the SCR reaction condition.24–26
In this study, we investigated the feasibility and performance of MOFs as a novel class of catalytic materials for the SCR of NOx with NH3 using the MIL-100(Fe) catalyst. Water and sulfur-resistance experiments were also performed to evaluate the durability of the catalyst in the presence of H2O and SO2. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was employed to explore the mechanism of NOx removal. To the best of our knowledge, there are no published reports describing the application of MOF materials for the SCR. This work may provide new insight into the design of efficient SCR catalysts with improved activity.
2. Experimental
2.1 Catalyst preparation
MIL-100(Fe) was prepared from the hydrothermal reaction of trimesic acid with metallic iron, HF, nitric acid and H2O.22 The molar composition of the reaction mixture was Fe0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,3,5-BTC:HF:HNO3:H2O = 1.0:0.67:2.0:0.6:277 (1,3,5-BTC = 1,3,5-benzenetricarboxylic or trimesic acid). The reactant mixture was loaded in a Teflon autoclave and maintained at 150 °C for 12 h. The as-synthesized MIL-100(Fe) was further purified by a two-step treatment using hot deionized water (80 °C) and ethanol (60 °C) for 3 h. Finally, the resulting light-orange solid was dried at 100 °C overnight.
:1,3,5-BTC:HF:HNO3:H2O = 1.0:0.67:2.0:0.6:277 (1,3,5-BTC = 1,3,5-benzenetricarboxylic or trimesic acid). The reactant mixture was loaded in a Teflon autoclave and maintained at 150 °C for 12 h. The as-synthesized MIL-100(Fe) was further purified by a two-step treatment using hot deionized water (80 °C) and ethanol (60 °C) for 3 h. Finally, the resulting light-orange solid was dried at 100 °C overnight.
The conventional catalyst (i.e., V2O5–WO3/TiO2 (V: ∼1%, W: ∼9%)) was prepared using the incipient wetness method.2 In a typical preparation, WO3 was added to a TiO2 support by impregnation with an aqueous solution of ammonium paratungstate complexed with citric acid, followed by drying and calcination at 550 °C for 3 h. Then, vanadium was added to the WO3/TiO2 system by impregnation starting from a water solution of ammonium metavanadate and oxalic acid, followed by drying and calcination at 550 °C for 2 h.
2.2 Characterization
Powder X-ray diffraction (XRD) patterns were recorded over the range of 3–40° on a Panalytical/Empyrean X-ray diffractometer with Cu Kα radiation. Infrared reflectance (IR) spectrums were recorded on a Bruker VERTEX 70 FTIR spectrometer equipped with DTGS detector in the range of 4000–600 cm−1 with the resolution of 4 cm−1. The morphology of MIL-100(Fe) was observed using a field-emission scanning electron microscope (SEM, Hitachi S-4800) operated at an accelerating voltage of 10 kV. The specific surface area was determined from a nitrogen adsorption isotherm using the Brunauer–Emmett–Teller (BET) model in Quantachrome SI. Thermogravimetric (TG) analysis was performed using a TG/DTA 6300 thermogravimetric analyzer from Seiko under an air stream (100 ml min−1) at a heating rate of 5 °C min−1 from 25 to 600 °C. X-ray photoelectron spectroscopy (XPS) was conducted on the surface analysis system (Thermal ESCALAB 250) using Al Kα radiation. The C 1s peak at 286.4 eV was used for the binding energy calibration.In situ DRIFTS measurements were performed on a Bruker VERTEX 70 FTIR spectrometer equipped with a MCT detector cooled by liquid nitrogen. An approximately 10 mg sample was finely ground and pressed into a self-supported wafer. The total gas flow was maintained at 100 ml min−1 by mass flow controllers. Prior to each experiment, the sample was pretreated at 250 °C for 10 h and cooled to room temperature in a flow of N2. At each temperature, the background spectrum was recorded in flowing N2 and automatically subtracted from the sample spectrum obtained at the same temperature. In the experiment, DRIFT spectra were recorded by accumulating 16 scans at a resolution of 4 cm−1.
2.3 Measurement of catalytic activity
SCR reaction tests were performed in a fixed-bed quartz tube reactor (ϕ = 9 mm) containing approximately 0.25 g of MIL-100(Fe) and 0.46 g of V2O5–WO3/TiO2. The MIL-100(Fe) catalyst was pretreated in N2 at 250°C for 10 h prior to the activity measurement. The feed gas consisted of 500 ppm NO, 500 ppm NH3, 4% O2, 500 ppm SO2 (when used), 5% H2O (when used), and N2 balance. The total flow rate was 315 ml min−1, and the gas hourly space velocity (GHSV) was 30000 h−1. The inlet and outlet NOx (NO plus NO2) concentrations were analyzed using a flow gas analyzer (ecom-J2KN, rbr Messtechnik GmbH Inc.). The concentration of NH3 was recorded through by NH3 analyzer (GXH-1050, Beijing), and the N2O concentration was monitored by a gas chromatograph (Shimadzu GC-14C) with a Porapak Q column. NOx conversion and N2 selectivity were calculated as follows:
 | (1) |
 | (2) |
The conversion rate was calculated from the conversion of NOx by using the pseudo-first-order kinetic equation as below.27
| r = −F/W × ln(1 − x) (mol NOx g cat−1 s−1) | (3) |
In the equation, F is the flow rate of NO (mol s−1), W is the weight of the catalyst (g) and x is NOx conversion (%). The conversion rate is affected by the reactant concentration, and in this equation the reaction orders of the reactant concentration with respect to NO, NH3 and H2O were first order, zero order and zero order, respectively. And the effect of O2 on the reaction rate is neglected due to its excessive concentration in the reaction.
3. Results and discussion
3.1 Physical and textural properties of MIL-100(Fe) catalyst
MIL-100(Fe) crystallizes in the cubic space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 227) with the cell parameter of a = 73.340 Å and V = 394481.1 Å3.22 Rietveld refinement plot for the XRD pattern of the as-synthesized MIL-100(Fe) was obtained by using the GSAS software.28 The results demonstrated that the peak positions matched well with that of the simulated diffraction pattern of MIL-100(Fe) in the range of 3 to 40 degree, which indicated that the crystalline state of the sample was consistent with MIL-100(Fe) (ESI, Fig. S1†). The SEM image indicated the size of the octahedral crystal, which was approximately 1.5 μm, as shown in Fig. 1a. The specific surface area and pore volumes determined by N2 physisorption were 2032 m2 g−1 and 1.12 cm3 g−1, respectively, greater than those of the conventional metal oxide catalyst. Therefore, MIL-100(Fe) could provide a high surface area, which is beneficial for the SCR reaction.29 In addition, the thermal stability of MIL-100(Fe) was studied by TG analysis from 25 to 600 °C in air, and the results are shown in Fig. S2.† A weight loss of 14.5% below 200 °C was due to the removal of physically and chemically adsorbed water.30 The loss observed at 200–300 °C was only 2.7%, which indicated that MIL-100(Fe) was thermally stable blow 300 °C. This thermal stability could ensure the applicability of the MIL-100(Fe) catalyst over this temperature range.
m (no. 227) with the cell parameter of a = 73.340 Å and V = 394481.1 Å3.22 Rietveld refinement plot for the XRD pattern of the as-synthesized MIL-100(Fe) was obtained by using the GSAS software.28 The results demonstrated that the peak positions matched well with that of the simulated diffraction pattern of MIL-100(Fe) in the range of 3 to 40 degree, which indicated that the crystalline state of the sample was consistent with MIL-100(Fe) (ESI, Fig. S1†). The SEM image indicated the size of the octahedral crystal, which was approximately 1.5 μm, as shown in Fig. 1a. The specific surface area and pore volumes determined by N2 physisorption were 2032 m2 g−1 and 1.12 cm3 g−1, respectively, greater than those of the conventional metal oxide catalyst. Therefore, MIL-100(Fe) could provide a high surface area, which is beneficial for the SCR reaction.29 In addition, the thermal stability of MIL-100(Fe) was studied by TG analysis from 25 to 600 °C in air, and the results are shown in Fig. S2.† A weight loss of 14.5% below 200 °C was due to the removal of physically and chemically adsorbed water.30 The loss observed at 200–300 °C was only 2.7%, which indicated that MIL-100(Fe) was thermally stable blow 300 °C. This thermal stability could ensure the applicability of the MIL-100(Fe) catalyst over this temperature range.
 | ||
| Fig. 1 SEM image of MIL-100(Fe). (a) MIL-100(Fe) before NH3-SCR activity test; (b) MIL-100(Fe) after 20 h NH3-SCR activity test at 250 °C; (c) MIL-100(Fe) after NH3-SCR durability test of resistance of SO2 and H2O at 250 °C. | ||
3.2 Catalytic performance
 | ||
| Fig. 2 NOx conversions over MIL-100(Fe) and V2O5–WO3/TiO2. Reaction condition: 500 ppm NO, 500 ppm NH3, 4% O2 and N2 balance, GHSV = 30000 h−1. | ||
Compared with the conventional catalysts reported in the ref. 31 and 32 the conversion rate of 0.74 × 10−6 mol NOx g cat−1 s−1 (calculated by eqn (3)) over MIL-100(Fe) at 200 °C is greatly higher than those of 0.09 × 10−6 mol NOx g cat−1 s−1 and 0.13 × 10−6 mol NOx g cat−1 s−1 over V2O5–WO3/TiO2 catalysts, which indicated MIL-100(Fe) catalyst was more efficient than that of the V2O5–WO3/TiO2 catalyst in the SCR reaction (Table 1).
However, the catalytic activity of MIL-100(Fe) decreased when the temperature was higher than 300 °C. The TG analysis results indicated that MIL-100(Fe) began to decompose, and the weight loss increased from 17.2 to 67.5% when the temperature increased from 300 to 405 °C. SEM images also indicated that MIL-100(Fe) collapsed after the SCR reaction at 325 and 350 °C (as shown in Fig. S3†). The XRD patterns of the samples used are shown in Fig. S4.† For the sample used at 300 °C, the typical MIL-100(Fe) phase was still the primary phase. In addition, the Fe2O3 phase was detected (2θ = 33.2 and 35.6° (PDF#33-0664)) for the two samples used at 325 and 350 °C. All of these results indicate that the decrease in NOx conversion may have been due to the gradual decomposition of the MIL-100(Fe) structure above 300 °C. In addition, the N2 selectivity was maintained at 100% over the temperature range of 100–260 °C. However, as the reaction temperature increased from 260 to 300 °C, the selectivity rapidly decreased, which generated increasing amounts of the unwanted N2O by-product (Fig. S5a†). For V2O5–WO3/TiO2 catalyst, only 95% of the selectivity to N2 was obtained when the high NOx conversion occurred at 350 °C (Fig. S5b†). The concentration of N2O over MIL-100(Fe) could not be detected in the active temperature range of 225–260 °C (NOx conversion > 90%), For V2O5–WO3/TiO2 catalyst, it increased from 11 ppm to 63 ppm in the temperature range of 300–400 °C. These results indicated MIL-100(Fe) exhibited the higher selectivity to N2 than the conventional V2O5–WO3/TiO2 catalyst at similar conversions in the SCR process.
 | ||
| Fig. 3 The stability tests of SCR reaction at 250 °C over MIL-100(Fe). Reaction condition: 500 ppm NO, 500 ppm NH3, 500 ppm SO2 (when used), 5% H2O (when used), 4% O2 and N2 balance, GHSV = 30000 h−1. | ||
SEM images of the catalysts used in the durability test are shown in Fig. 1. The images indicate that the octahedral geometrical shape of the catalyst was retained after the durability test with and without SO2 and H2O (Fig. 1b and c). The XRD patterns of the catalysts used are shown in Fig. 4a. As the inset of Fig. 4a displayed, it was clearly demonstrated the MIL-100(Fe) catalyst were stable in the SCR reaction even with the presence of SO2 and H2O because all characteristic peaks of the used catalysts were still observed and the position was not changed compared with the as-synthesized MIL-100(Fe). Furthermore, the patterns of IR spectrum of MIL-100(Fe) catalysts before and after the durability tests were displaced in Fig. 4b. The characteristic peaks24,33 (ν(C–O) at 1634 and 1378 cm−1 and ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) at 1578 and 1453 cm−1) of the used catalysts were similar with the catalyst MIL-100(Fe) before durability test, which was in accord with the results of XRD patterns of the as-synthesized and used MIL-100(Fe) catalysts. The positive peak (curve d) appeared at 1085 cm−1 could be attributed to the deposited sulfate34 on the surface of MIL-100(Fe) catalyst after durability test with SO2 and H2O.
C) at 1578 and 1453 cm−1) of the used catalysts were similar with the catalyst MIL-100(Fe) before durability test, which was in accord with the results of XRD patterns of the as-synthesized and used MIL-100(Fe) catalysts. The positive peak (curve d) appeared at 1085 cm−1 could be attributed to the deposited sulfate34 on the surface of MIL-100(Fe) catalyst after durability test with SO2 and H2O.
 | ||
| Fig. 4 (a) Comparison of power X-ray diffraction patterns of various MIL-100(Fe) catalysts: (a) before activity test; (b) after durability test without SO2 and H2O; (c) after durability test with SO2 and H2O; (b) comparisons of IR spectrum of various MIL-100(Fe) catalyst: (a) before activity test; (b) after durability test without SO2 and H2O; (c) after durability test with SO2 and H2O; (d) difference spectra obtained by spectra a subtracting spectra c.; (c) XPS spectra of S 2p: (a) MIL-100(Fe) before durability test; (b) as-synthesized MIL-100(Fe) after durability test with SO2 and H2O. | ||
The oxidation states of surface elements on MIL-100(Fe) catalysts before and after durability test were characterized using XPS. As shown in Fig. 4c, no S 2p band was observed on MIL-100(Fe) catalyst before durability test. After durability test with SO2 and H2O, an evident band at 186.6 eV attributed to S 2p35 was observed and the S content was calculated to be 1.15% (shown in Table 2), implying the formation of sulfur-containing species on the catalyst surface. Moreover, the Fe content in the used catalyst declined compared with the fresh MIL-100(Fe) catalyst. The reason could be attributed to the coverage of the formed sulfate on the iron sites. Furthermore, the C/O ratio of the used MIL-100(Fe) catalyst was similar with that of the fresh MIL-100(Fe). The result indicated the effect of organic coordination (i.e. the residual trimesic acid) in MIL-100(Fe) on the SCR catalytic activity could be ignored.
| Catalysts | Fe | C | O | F | S |
|---|---|---|---|---|---|
| a a, MIL-100(Fe) before durability test; b, MIL-100(Fe) after durability test with SO2 and H2O. | |||||
| a | 4.59 | 65.85 | 29.01 | 0.55 | — |
| b | 3.47 | 65.98 | 29.26 | 0.14 | 1.15 |
To elucidate the surface acidity on fresh and used MIL-100(Fe), in situ DRIFT spectra of NH3 adsorption were recorded. Prior to the test, the catalysts (0.01 g) were exposed in NH3/N2 (500 ppm) for 1 h after treating with N2 for 30 min to evacuate the impurity at 250 °C, and followed by purging with N2 for 30 min at 250 °C and the results were displayed in Fig. S6.† The bands appeared at 1735, 1662 and 1455 cm−1 were attributed to asymmetric and symmetric bending vibrations of NH4+ species on Brønsted acid sites.36,37 The bands displayed at 1603, 1296 and 1151 cm−1 were assigned to coordinated NH3 on Lewis acid sites.38 It is well known the relative quantity of Brønsted acid sites (1735 and 1662 cm−1 assigned to δs(NH4+)) and Lewis acid sites (1296 and 1151 cm−1 assigned to δs(NH3)) on surface of MIL-100(Fe) catalyst could be calculated by the corresponding integral characteristic peak area. According to Fig. S6b,† the results indicated both the intensity of B and L acid sites of the used MIL-100(Fe) decreased compared with the fresh MIL-100(Fe). The deposited sulfate on the active sites could prevent the adsorption and activation of gaseous NH3, which induced the decrease of quantity of acid sites on MIL-100(Fe).
3.3 In situ DRIFT studies
To investigate the catalytic reaction mechanism, DRIFTS experiments were performed to identify the species absorbed on the catalyst surface and deduce the surface reaction under actual reaction conditions. | ||
| Fig. 5 DRIFT spectra of MIL-100(Fe) exposed to NH3 at various temperatures. Reaction condition: [NH3] = 500 ppm, and N2 balance. | ||
 | (4) |
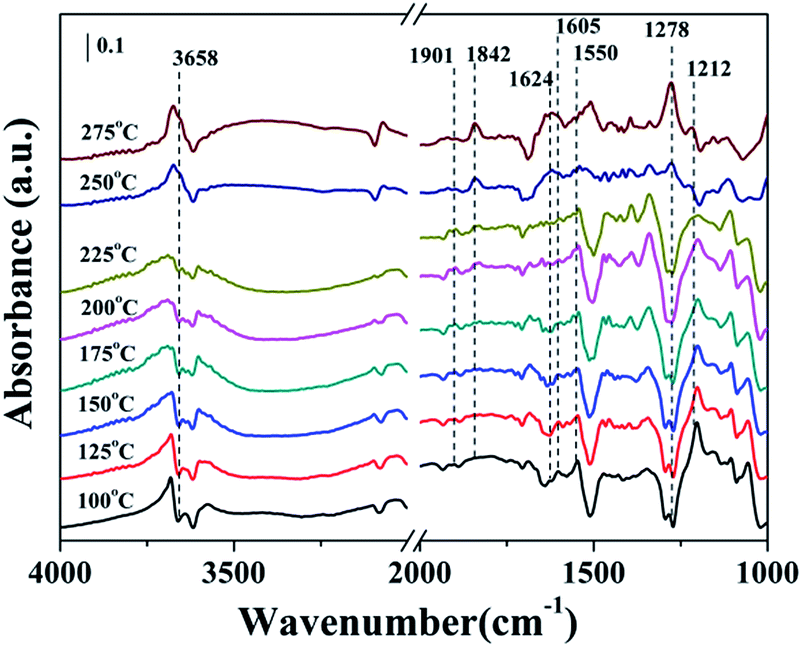 | ||
| Fig. 6 DRIFT spectra of MIL-100(Fe) exposed to NO + O2 at various temperatures. Reaction condition: [NO] = 500 ppm, [O2] = 4% and N2 balance. | ||
 | ||
| Fig. 7 DRIFT spectra of MIL-100(Fe) exposed to NOx after NH3 adsorption and purged with N2 at 250 °C for various times. Reaction condition: [NH3] = 500 ppm, [NO] = 500 ppm, [O2] = 4% and N2 balance. | ||
 | ||
| Fig. 8 DRIFT spectra of MIL-100(Fe) exposed to NH3 after NO and O2 co-adsorption and purged with N2 at 250 °C for various times. Reaction condition: [NH3] = 500 ppm, [NO] = 500 ppm, [O2] = 4% and N2 balance. | ||
 | ||
| Fig. 9 DRIFT spectra of MIL-100(Fe) exposed to the SCR reaction at 250 °C for various times. Reaction condition: [NH3] = 500 ppm, [NOx] = 500 ppm, [O2] = 4% and N2 balance. | ||
The DRIFT results for NH3 adsorption indicated that both Brønsted and Lewis acid sites existed on the surface of MIL-100(Fe), and NH3 was primarily adsorbed on MIL-100(Fe) in the form of ionic NH4+ and coordinated NH3. In addition, after the introduction of NO + O2, the disappearance of the NH4+ species indicated that ionic NH4+ may have been the primary reactive ammonia species adsorbed on MIL-100(Fe) during the reaction.
Because NO was easily oxidized by O2 on Fe3+,47 NO was adsorbed on the iron sites to form NO2 species (as described in Section 3.3.2). Then, the reactive NO2 could react with the neighboring NH4+ on the iron sites to form N2. Therefore, we propose that a Langmuir–Hinshelwood (L–H) mechanism was dominant in the SCR reaction on MIL-100(Fe), as described by the following reaction scheme.
| NH3(g) → NH3(a) → NH+4(a) → NH+4(a) + NO2(a) → N2(g) + H2O(g) | (5) |
During this process, Fe(III) CUS could be reduced to Fe(II) CUS in the oxidation step of adsorbed NO to NO2, and then, Fe(II) was reoxidized to Fe(III) by O2 to complete the redox cycle.
 | (6) |
The coordinated NH3 was considered to be less reactive due to the high activation energy required to form NH2 species.48 However, based on the results presented in Section 3.3.1, the coordinated NH3 adsorbed on MIL-100(Fe) could undergo oxidative dehydrogenation to form NH2 species. In addition, the NH2 species could react with NO (shown in Fig. 5), suggesting that the SCR reaction on MIL-100(Fe) could also occur via the Eley–Rideal (E–R) mechanism.
| NH3(g) → NH3(a) → NH2(a) → NH2(a) + NO(g) → N2(g) + H2O(g) | (7) |
The stable structure of the MIL-100(Fe) catalyst used in the durability test indicated that SO2 did not easily react with the iron active sites. In addition, the absence of sulfate on the surface of used MIL-100(Fe) catalyst after durability test suggested that the porous structure of MIL-100(Fe) may prevent the deposition of sulfate. Therefore, a high conversion of NOx could be recovered by eliminating SO2. The declined NOx conversion may be attributed to the competitive adsorption between SO2 and reactants.
4. Conclusions
The porous MOF MIL-100(Fe) catalyst was observed to be an excellent catalyst and exhibited better performance in the SCR of NOx with NH3 at lower temperatures (245–300 °C) than those required by the conventional V2O5–WO3/TiO2 catalyst. In addition, the MIL-100(Fe) catalyst exhibited satisfactory stability in the presence of H2O and SO2 and good self-regeneration capability for the SCR of NOx after eliminating H2O and SO2. XRD, IR and SEM analyses indicated that the structure of the catalyst used in the durability test was the same as that of the fresh sample, suggesting that MIL-100(Fe) was stable during the SCR process below 300 °C. Based on the DRIFT results, two reaction pathways were proposed. The reaction between the NH4+ species and the adsorbed NO2 species on Fe(II) CUS was the primary reaction pathway, which is consistent with the L–H mechanism. The SCR process also followed the E–R mechanism, through which the amide species reacted with gaseous NO. The valance change of Fe promoted the formation of adsorbed NO2 species, which were beneficial for the SCR reaction. This study provides insight that may benefit future investigations of MOFs as high-efficiency catalysts in the SCR process. In addition, this work supports the further study of high NOx conversion over MOFs at low temperature.Acknowledgements
This work was supported by the National Basic Research Program of China (2011CB936002) and the National Natural Science Foundation of China (no. 21277015).Notes and references
- L. J. Alemany, F. Berti, G. Busca, G. Ramis, D. Robba, G. P. Toledo and M. Trombetta, Appl. Catal., B, 1996, 10, 299–311 CrossRef CAS.
- L. Lietti, I. Nova and P. Forzatti, Top. Catal., 2000, 11/12, 111–122 CrossRef CAS.
- L. Lietti, Appl. Catal., B, 1996, 10, 281–297 CrossRef CAS.
- L. Chen, J. Li and M. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- J. Xue, X. Wang, G. Qi, J. Wang, M. Shen and W. Li, J. Catal., 2013, 297, 56–64 CrossRef CAS PubMed.
- P. R. Ettireddy, N. Ettireddy, T. Boningari, R. Pardemann and P. G. Smirniotis, J. Catal., 2012, 292, 53–63 CrossRef CAS PubMed.
- Y. J. Kim, H. J. Kwon, I. Heo, I. Nam, B. K. Cho, J. W. Choung, M. Cha and G. K. Yeo, Appl. Catal., B, 2012, 126, 9–21 CrossRef CAS PubMed.
- J. Park, H. J. Park, J. H. Baik, I. Nam, C. Shin, J. Lee, B. K. Cho and S. H. Oh, J. Catal., 2006, 240, 47–57 CrossRef CAS PubMed.
- T. Boningari, R. Koirala and P. G. Smirniotis, Appl. Catal., B, 2013, 140–141, 289–298 CrossRef CAS PubMed.
- W. S. Kijlstra, M. Biervliet, E. K. Poels and A. Bliek, Appl. Catal., B, 1998, 16, 327–337 CrossRef.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- L. S. Cheng, R. T. Yang and N. Chen, J. Catal., 1996, 164, 70–81 CrossRef CAS.
- L. Y. Kong, Z. H. Zhang, H. F. Zhu, H. Kawaguchi, T. Okamura, M. Doi, Q. Chu, W. Y. Sun and N. Ueyama, Angew. Chem., Int. Ed., 2005, 44, 4352–4355 CrossRef CAS PubMed.
- X. S. Wang, S. Q. Ma, D. F. Sun, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 16474–16475 CrossRef CAS PubMed.
- R. Q. Zou, H. Sakurai, S. Han, R. Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402–8403 CrossRef CAS PubMed.
- S. Han, Y. Huang, T. Watanabe, S. Nair, K. S. Walton, D. S. Sholl and J. C. Meredith, Microporous Mesoporous Mater., 2013, 173, 86–91 CrossRef CAS PubMed.
- N. Apostolescu, B. Geiger, K. Hizbullah, M. T. Jan, S. Kureti, D. Reichert, F. Schott and W. Weisweiler, Appl. Catal., B, 2006, 62, 104–114 CrossRef CAS PubMed.
- M. Iwasaki, K. Yamazaki, K. Banno and H. Shinjoh, J. Catal., 2008, 260, 205–216 CrossRef CAS PubMed.
- F. Liu, H. He and C. Zhang, Chem. Commun., 2008, 2043–2045 RSC.
- P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC.
- J. W. Yoon, Y. K. Seo, Y. K. Hwang, J. S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J. M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS PubMed.
- S. Wuttke, P. Bazin, A. Vimont, C. Serre, Y. Seo, Y. K. Hwang, J. Chang, G. Férey and M. Daturi, Chem.–Eur. J., 2012, 18, 11959–11967 CrossRef CAS PubMed.
- C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2011, 21, 2108–2117 CrossRef CAS.
- E. Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174–10181 CrossRef PubMed.
- P. Sazama, B. Wichterlová, E. Tábor, P. Šťastný, N. K. Sathu, Z. Sobalík, J. Dědček, Š. Sklenák, P. Klein and A. Vondrová, J. Catal., 2014, 312, 123–138 CrossRef CAS PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- X. Fan, F. Qiu, H. S. Yang, W. Tian, T. F. Hou and X. B. Zhang, Catal. Commun., 2011, 12, 1298–1301 CrossRef CAS PubMed.
- R. Canioni, C. R. Marchal, F. Sécheresse, P. Horcajada, C. Serre, M. Hardi-Dan, G. Férey, J. M. Grenèche, F. Lefebvre, J. S. Chang, Y. K. Hwang, O. Lebedev, S. Turner and G. V. Tendeloo, J. Mater. Chem., 2011, 21, 1226–1233 RSC.
- S. Yang, C. Wang, L. Ma, Y. Peng, Z. Qu, N. Yan, J. Chen, H. Chang and J. Li, Catal. Sci. Technol., 2013, 3, 161–168 CAS.
- P. Balle, B. Geiger and S. Kureti, Appl. Catal., B, 2009, 85, 109–119 CrossRef CAS PubMed.
- M. Haouas, C. Volkringer, T. Loiseau, G. Férey and F. Taulelle, J. Phys. Chem. C, 2011, 115, 17934–17944 CAS.
- T. Yamaguchi, T. Jin and K. Tanabe, J. Phys. Chem., 1986, 90, 3148–3152 CrossRef CAS.
- M. S. Maqbool, A. K. Pullur and H. P. Ha, Appl. Catal., B, 2014, 152–153, 28–37 CrossRef CAS PubMed.
- S. D. Lin, A. C. Gluhoi and B. E. Nieuwenhuys, Catal. Today, 2004, 90, 3–14 CrossRef CAS PubMed.
- B. Jiang, Z. Li and S. Lee, Chem.–Eur. J., 2013, 225, 52–58 CAS.
- G. Ramis, L. Yi and G. Busca, Catal. Today, 1996, 28, 373–380 CrossRef CAS.
- J. Y. Luo, H. Oh, C. Henry and W. Epling, Appl. Catal., B, 2012, 123–124, 296–305 CrossRef CAS PubMed.
- W. S. Kijlstra, D. S. Brands, E. K. Poels and A. Bliek, J. Catal., 1997, 171, 219–230 CrossRef CAS.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS.
- M. A. Larrubia, G. Ramis and G. Busca, Appl. Catal., B, 2001, 30, 101–110 CrossRef CAS.
- M. A. Larrubia, G. Ramis and G. Busca, Appl. Catal., B, 2000, 27, L145–L151 CrossRef CAS.
- W. S. Kijlstra, D. S. Brands, E. K. Poels and A. Bliek, J. Catal., 1997, 171, 208–218 CrossRef CAS.
- M. Machida, M. Uto, D. Kurogi and T. Kijima, Chem. Mater., 2000, 12, 3158–3164 CrossRef CAS.
- S. Wuttke, P. Bazin, A. Vimont, C. Serre, Y. Seo, Y. K. Hwang, J. Chang, G. Férey and M. Daturi, Chem.–Eur. J., 2012, 18, 11959–11967 CrossRef CAS PubMed.
- R. Q. Long and R. T. Yang, J. Catal., 2002, 207, 224–231 CrossRef CAS.
- G. Qi and R. T. Yang, J. Phys. Chem. B, 2004, 108, 15738–15747 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07028c |
| This journal is © The Royal Society of Chemistry 2014 |