DOI:
10.1039/C4RA06972B
(Paper)
RSC Adv., 2014,
4, 34443-34447
On-chip direct freezing and thawing of mammalian cells†
Received
11th July 2014
, Accepted 28th July 2014
First published on 28th July 2014
Abstract
Here we present a simple protocol for directly freezing and thawing mammalian cells on a PDMS–glass chip, which enables cell storage at −80 °C on a chip for several days to several months. Two different sizes of straight channels were used (channel I: 4 cm length, 2 mm width, 100 μm height; and channel II: 1 cm length, 900 μm width, 60 μm height). The cell suspension (cell pellet resuspended in freezing medium, which was composed of dimethylsulfoxide (DMSO), fetal bovine serum (FBS) and culture medium) was injected into the microchannel, and the chip was packed and put into the centrifuge tube filled with isopropyl alcohol. The centrifuge tubes were maintained at −80 °C in a cryogenic refrigerator until further use. We demonstrated that HUVECs, 3T3, SKBR3, and HepG2 cells were successfully thawed and grew in the microchannel after 10-days to 1-year cryopreservation by using this protocol.
Introduction
Cell freezing and thawing are basic techniques used for mammalian cell culture. Cryopreservation prevents the need for continuous cell cultures. The procedure can also reduce the risk of contamination, minimize genetic and morphologic changes, and maintain a consistent cell passage number, which are especially valuable for cells of limited life spans. Cells can be frozen at −70 °C to −90 °C with cryoprotection by using a slow cooling process and then stored in a liquid nitrogen freezer for long term preservation. Cells can be revived from the frozen state to 37 °C as rapidly as possible to reach a higher cell survival rate.1–4 Dimethylsulfoxide (DMSO) is one of the most widely used cryoprotection agents, which can help prevent cell dehydration during freezing.5
In the slow cooling process, a cooling rate of −1 °C to −3 °C per minute is recommended for most mammalian cells.6 Instead of using homemade devices, such as plastic boxes packed with cotton, Nalgene Mr Frosty Cryo 1 °C Freezing Containers have become a convenient choice. The Mr Frosty container, filled with isopropyl alcohol and cell cryogenic storage vials placed into holes, can be directly placed into a −80 °C cryogenic refrigerator. The cell cryogenic storage vial is usually made of polypropylene.
Microfluidic devices have recently become a versatile platform for applications in cell-based assays. These devices offer a number of distinct advantages over traditional research platforms. Such advantages include low cost, reduced consumption, and high throughput. When cell-based experiments, such as cell transfection,7,8 cell apoptosis,9 chemotaxis assays,10 and cell response to mechanical forces,11,12 are performed on microfluidic chips, the general process of cell preparation could be described as follows. First, cells stored in liquid nitrogen are revived and cultured in tissue culture dishes or flasks. Second, the cells are dissociated from the culture vessel using trypsin–EDTA after reaching confluence. Then, the cell suspension is adjusted to a proper density and injected into the microchannel. When cells in channel attain confluence, cell-based experiments are initiated to perform on chip. Preparation usually takes several days up even one week to two weeks before the cells cultured on chip are ready for the main experiments. Therefore, directly freezing and thawing cells on chip is beneficial for lab-on-a-chip technology and microchip-based life science.
Microfluidic technology was used to minimize osmotic shock to cells during cryopreservation.13 In addition, several reports described the on-chip cryopreservation of living cells.14,15 However, the fabrications of these platforms are more complex than those of the PDMS–glass chips. PDMS has been proven to be safe to cells, and PDMS–glass chips are commonly used for on-chip cell-based experiments. Small amounts of human spermatozoa were successfully frozen and thawed on PDMS chips.16 In this paper, we propose an on-chip direct freezing and thawing protocol for mammalian cells. Small amount of cells can be stored on PDMS–glass chip for several days to several months. This protocol is a simple method that only uses common materials found in a biology laboratory. The consumables of valuable cells, culture medium and the preparation time of the on-chip cell-based experiment can be remarkably reduced. The subsequent cell manipulations and treatments can be performed by using the thawed cells on the chip. Considering these aspects, we believe that this strategy could become a fundamental technique for an integrated lab on a chip. It also provides an avenue for cell storage and transfer.
Materials and methods
Fabrication of the PDMS–glass chip
Two different sizes of straight channels were used. Channel I is 4 cm long, 2 mm wide and 100 μm high, and channel II is 1 cm long, 900 μm wide and 60 μm high. The microchannel structure was fabricated in PDMS (Sylgard 184, Dow Corning) by rapid prototyping and replica molding techniques.17 Briefly, the negative relief of PDMS was formed by curing the prepolymer on a silanized Si master with a positive relief of the channels formed in photoresist (SU-8 2050/2075, MicroChem) on the surface. Inlets were drilled using a blunted and beveled syringe needle. Finally, oxygen plasma treatment was performed by using a Plasma Cleaner (PDC-32G, Harrick Scientific Products, Inc.) to bond the PDMS replica to a clean slide glass, finally forming a complete microfluidic chip.
On-chip freezing of mammalian cells
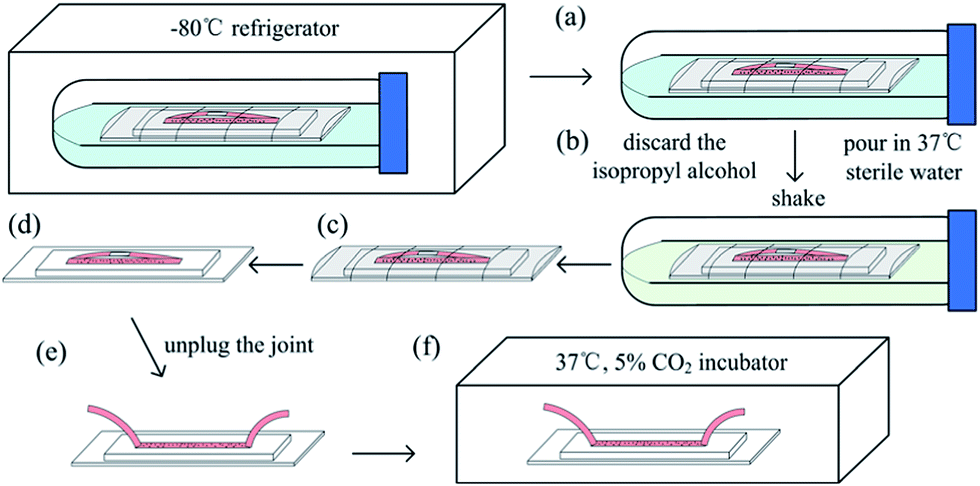
Prior to cell seeding, all the chips, tubes and joints were autoclaved for 20 min at 121 °C and a 50 μg mL−1 human fibronectin (Millipore) solution was injected into the channels to promote cell adhesion. The freezing medium was composed of 5% DMSO (Sigma), 50% fetal bovine serum (FBS, Gibco) and 45% dulbecco's modified eagle medium (DMEM, Gibco) for HepG2, 3T3 and SKBR3 cells or 45% endothelial cell medium (ECM, ScienCell) for HUVECs. The freezing protocol and chip pictures are shown in Fig. 1 and 2. Briefly, cells cultured in flasks were removed with trypsin–EDTA solution at approximately 90% confluence and then resuspended in freezing medium (Fig. 1a). The number of cells was adjusted to 5 × 106 to 5 × 107 mL−1. Thereafter, the cell suspension was carefully injected into the channel (Fig. 1b and 2a), and the inlet and outlet tubes were connected by a joint (Fig. 1c and 2b). The chip was packed by parafilm (Fig. 1d and 2c) or placed in a small ziplock bag. The packed chip was put into a 50 mL centrifuge tube filled with approximately 25 mL of isopropyl alcohol (Fig. 1e and 2d). The bottom of the chip must be in contact with the liquid level of the isopropyl alcohol. Finally, the 50 mL centrifuge tube was placed horizontally in the −80 °C refrigerator until further use (Fig. 1f).
 |
| | Fig. 1 On-chip cell freezing protocol. (a) Cells were resuspended in the freezing medium, (b) cell suspension was injected into the microchannel, (c) the inlet and outlet tubes were connected, (d) the chip was packed, (e) the chip was put into the centrifuge tube filled with isopropyl alcohol, and (f) the centrifuge tube was placed horizontally in the −80 °C refrigerator. | |
 |
| | Fig. 2 Photos of the chip and the freezing unit. (a) Cell suspension was injected into the microchannel, (b) the inlet and outlet tubes were connected by a joint, (c) the chip was packed by parafilm, and (d) the packed chip was put in a 50 mL centrifuge tube, which filled with about 25 mL of isopropyl alcohol. | |
On-chip thawing of mammalian cells
The on-chip thawing protocol is shown in Fig. 3. First, the 50 mL centrifuge tube (with the chip in) was removed from the refrigerator (Fig. 3a). The cap was twisted off, the isopropyl alcohol was discarded, and 37 °C sterile water was poured in (Fig. 3b). The tube was shaken quickly, and the chip was extracted (Fig. 3c). The parafilm was removed (Fig. 3d), the joint was unplugged (Fig. 3e), and the chip was put in the humidified incubator at 37 °C with 5% CO2 (Fig. 3f). The freezing medium in the microchannel and the inlet and outlet tubes was replaced with fresh cell culture medium after approximately 8 h when the cells had adhered to the glass surface. The culture is continued in the incubator and the medium was replaced every 10 to 12 h. Cells were inspected and recorded by using a microscope (Ti-s, Nikon) and a CCD camera (Ds-Ri1, Nikon).
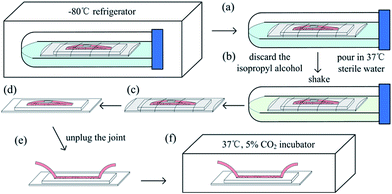 |
| | Fig. 3 On-chip cell thawing protocol. (a) The centrifuge tube was removed from the refrigerator, (b) the isopropyl alcohol was discarded and 37 °C sterile water was poured in, (c) the chip was extracted, (d) the parafilm was removed, (e) the joint was unplug, and (f) the chip was placed in the 37 °C, 5% CO2 incubator. | |
Live-dead assay
After 15 days in the freezer and 8 h after thawing, the 3T3 cells and HUVECs were analyzed using the LIVE/DEAD Viability/Cytotoxicity Assay Kit for mammalian cells (MP 03224, Invitrogen) and fluorescent microscopy to determine their viabilities. The kit contains two fluorescent stains, namely, calcein AM and ethidium homodimer-1 (EthD-1), which were used as their recommended concentrations of 2 μM calcein AM and 4 μM EthD-1, respectively. The cells in the channel were first washed by DPBS, and then 100 μL of combined stain solution was infused into the channel. After 20 min of incubation, the cells were washed by DPBS and then viewed with a fluorescence microscope (Ti-s, Nikon). Green and red fluorescence images were captured with a CCD camera (Ds-Ri1, Nikon) and overlaid at 50% opacity.
Cell adhesion number
We compared the number of adhering 3T3 cells frozen on chip between 20 days and four months, and the number of adhering HUVECs frozen on chip between 20 days and 1 year, respectively. Cell culture medium was infused into the channel to replace the freezing medium at 8 h after thawing. The number of cells remaining in the channel was calculated. Cell numbers were counted from four randomly captured microscope field images. Data are expressed as means ± standard deviations (more than three replicates were conducted in each experiment).
Results and discussion
We froze several types of mammalian cells in channel I and II, and then thawed these cells after several days to test this protocol. The results of direct thawing and subsequent cultivation of the cells in microchannels are shown in Fig. 4, 5 and S1–S4 (see ESI†).
 |
| | Fig. 4 Images of the 3T3 cells in channel I, thawed after 20 days cryopreservation, (a and b) 8 h after thawing, (c and d) 24 h after thawing, (e and f) 48 h after thawing. The objective magnifications were 4× (left) and 10× (right). | |
 |
| | Fig. 5 Images of the HUVEC cells in channel I, thawing after 20 days cryopreservation, (a and b) 8 h after thawing, (c and d) 24 h after thawing, (e and f) 48 h after thawing. The objective magnifications were 4× (left) and 10× (right). | |
The 3T3 cells and HUVECs directly frozen in channel I were thawed after 20 days in storage. Cell culture medium was infused into the channel to replace the freezing medium 8 h after thawing, and the adhering cells are shown in Fig. 4(a), (b) and 5(a), (b) with objective magnifications of 4× for (a) and 10× for (b). The cells have spread and grown during the subsequent 16 h, as shown in Fig. 4(c), 4(d) and 5(c), (d). Two days after thawing, cells in channels have reached 80% to 90% confluence, as shown in Fig. 4(e), (f) and 5(e), (f). More results are shown in the ESI.† HepG2 cells, frozen for 15 days in channel I, are shown in Fig. S1.† SKBR3 cells, frozen for 10 days in channel II, are shown in Fig. S2.†
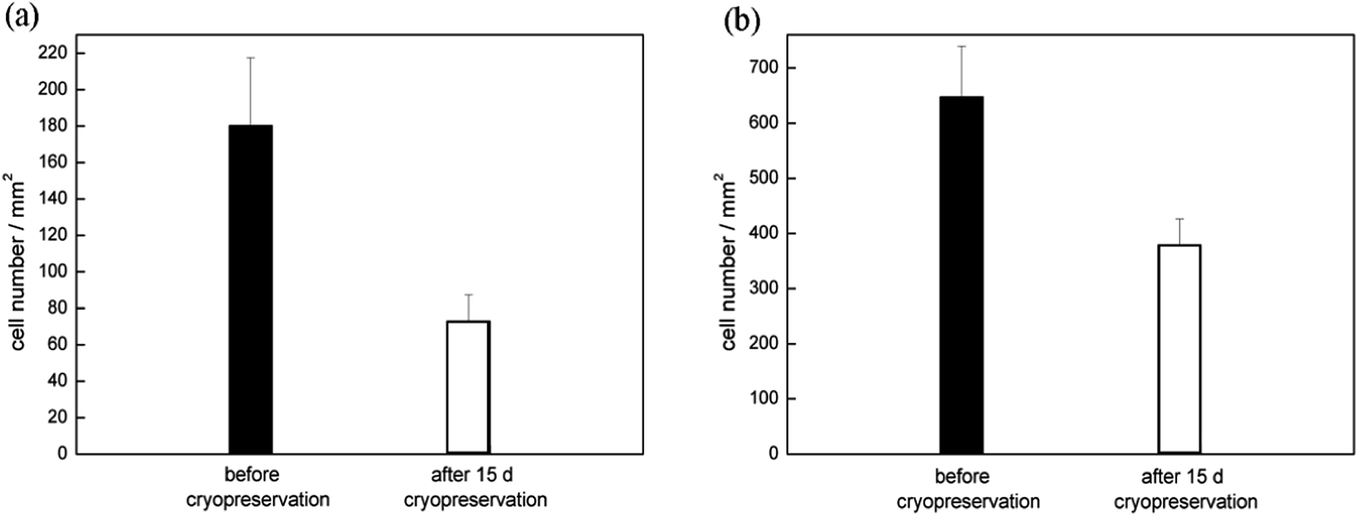
To assess the effect of direct freezing and thawing on chip on cell viability, we performed live-dead assays on 3T3 cells and HUVECs. Calcein-AM producing green fluorescence was used as the live stain. Live cells are indicated by the conversion of calcein AM to calcein by intracellular esterases. Meanwhile, EthD-1 enters cells with damaged membranes and produces red fluorescence in dead cells. Before store in cryogenic refrigerator, the 3T3 cells and HUVECs in channels were captured and shown in Fig. 6(a) and (b). Cells were frozen on chip for 15 days and then thawed with subsequent 8 h culture in channels. Following exposure of cells to live/dead stains for 20 min of incubation, bright field images were captured and shown in Fig. 6(c) and (d), and fluorescence images were captured, overlaid at 50% opacity, and shown in Fig. 6(e) and (f). The number of cells of the green fluorescence image was counted and compared with that of the cells in the bright field image in the same field of view. We found that the vast majority of the adhering cells are stained green (98.4 ± 1.0% of 3T3 cells and 99.2 ± 0.2% of HUVECs), whereas only a few cells are stained red. We also compared the number of cells of the green fluorescence image with the number of the cells in channels before cryopreservation. The results were shown in Fig. 7.
 |
| | Fig. 6 Cells in channel I, 3T3 cells (left) and HUVECs (right), (a and b) before cryopreservation, (c to f): live/dead stain results, 8 h after thawing, green for live cells and red for dead cells, thawing after 15 days cryopreservation. (c and d) Bright field images, (e and f) fluorescence images, overlaid at 50% opacity. The objective magnifications were 4×. Scale bar = 500 μm. | |
 |
| | Fig. 7 The number of cells in channels, (■) before cryopreservation, (□) the number of live cells, counted at 8 h after thawing, store for 15 days, (a) 3T3 cells, (b) HUVECs. | |
Moreover, we successfully thawed 3T3 cells after four months of storage and HUVECs after 1 year of storage on chip. The results are shown in Fig. S3 and S4.† Although the number of adhering cells was less than that in the channel after 20 days of storage (see Fig. 4(a), (b) and 5(a) and (b)), some cells remained alive and grew in the channel during the process of cultivation, as shown in Fig. S3(c) to S3(f), and S4(c) to S4(f).† The adhering cell number comparison at 8 h after thawing is shown in Fig. 8. The number of live cells decreased with longer freezing periods. Therefore, we recommend the use of cells frozen on chip as early as possible.
 |
| | Fig. 8 The number of adhering cells in channels, counted at 8 h after thawing, (a) 20 days and four months of freezing of 3T3 cells, (b) 20 days and 1 year of freezing of HUVECs. | |
Conclusions
Here we present a protocol for the direct freezing and thawing of mammalian cells on PDMS–glass chips, which enables the storage of cells for several days up to months. By using this protocol, we can keep a small amount of cells in microchannel in a −80 °C cryogenic refrigerator and revive them when needed. We tested several mammalian cells, such as cancer cells (SKBR3 and HepG2), endothelial cell (HUVECs) and fibroblast cell (3T3). Moreover, different freezing periods (from 10 days to 1 year) and channel sizes were studied. The advantage of the proposed method is lies in its simplicity and the use of common materials found in a biology laboratory. Directly freezing and thawing cells on chip, instead of thawing and culturing cells in flask and transferring them in micro channels, will remarkably reduces the consumables of valuable cells, culture medium, and the preparation time for on-chip cell-based experiments. This strategy would be beneficial for not only cell storage for long-term period, but also transferring cells.
Acknowledgements
This work was supported by the National Basic Research Program of China (no. 2011CB933102), Key Research Program of the Chinese Academy of Sciences (no. KJZD-EW-TZ-L03-6), and Key Laboratory of Cryogenics, TIPC, CAS (no. CRYOQN201306).
Notes and references
- R. I. Freshney, Culture of Animal Cells. A Manual of Basic Technique, Wiley, New York, 3rd edn, 1994 Search PubMed.
- R. J. Hay, Methods Cell Sci., 1978, 4, 787–790 Search PubMed.
- C. B. Schroy and P. Todd, Methods Cell Sci., 1976, 2, 309–310 Search PubMed.
- J. Shannon and M. Macy, Tissue Culture: Methods and Applications. ed. P. F. Kruse and M. K. Patterson Jr, Academic Press, New York, 1973, pp. 712–718 Search PubMed.
- N. C. Santos, J. Figueira-Coelho, J. Martins-Silva and C. Saldanha, Biochem. Pharmacol., 2003, 65, 1035–1041 CrossRef CAS.
- P. Mazur, Am. J. Physiol.: Cell Physiol., 1984, 247, C125–C142 CAS.
- L. Li, Y. Nie, D. Ye and G. Cai, Lab Chip, 2009, 9, 2230–2233 RSC.
- L. Li, Y. Nie, X. Shi, H. Wu, D. Ye and H. Chen, Biomicrofluidics, 2011, 5, 036503 CrossRef PubMed.
- C. G. Yang, Y. F. Wu, Z. R. Xu and J. H. Wang, Lab Chip, 2011, 11, 3305–3312 RSC.
- N. L. Jeon, H. Baskaran, S. K. W. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CrossRef CAS PubMed.
- E. Tkachenko, E. Gutierrez, M. H. Ginsberg and A. Groisman, Lab Chip, 2009, 9, 1085–1095 RSC.
- L. Li, Y. Yang, X. Shi, H. Wu, H. Chen and J. Liu, Microfluid. Nanofluid., 2014, 16(6), 1089–1096 CrossRef CAS.
- Y. S. Song, S. Moon, L. Hulli, S. K. Hasan, E. Kayaalp and U. Demirci, Lab Chip, 2009, 9, 1874–1881 RSC.
- E. Berthier, D. J. Guckenberger, P. Cavnar, A. Huttenlocher, N. P. Keller and D. J. Beebe, Lab Chip, 2013, 13, 424–431 RSC.
- S. Li, W. Liu and L. Lin, JALA, 2010, 15, 99–106 CrossRef CAS.
- Y. Zou, T. Yin, S. Chen, J. Yang and W. Huang, PLoS One, 2013, 8, e61593 CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06972b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.