DOI:
10.1039/C4RA06917J
(Paper)
RSC Adv., 2014,
4, 55853-55866
Quantitative structure–activity relationships and design of thymine-like inhibitors of thymidine monophosphate kinase of Mycobacterium tuberculosis with favourable pharmacokinetic profiles
Received
10th July 2014
, Accepted 16th October 2014
First published on 17th October 2014
Abstract
We have designed new potent inhibitors of thymidine monophosphate kinase of Mycobacterium tuberculosis (TMPKmt) using structure-based molecular design. Three-dimensional (3D) models of TMPKmt–inhibitor complexes were prepared by in situ modification of the crystal structure of TMPKmt co-crystallized with the natural substrate deoxythymidine monophosphate (dTMP) (PDB entry code: 1G3U) and a training set of 20 thymine derivatives bearing an aliphatic or aromatic group attached through a spacer (THMDs) with known inhibitory potencies. A QSAR model was elaborated for the training set THMDs and a linear correlation was established between the computed free energies of THMD binding and observed enzyme inhibition constants (Kexpi). Validation of this QSAR model was performed with 3D-QSAR pharmacophore generation (PH4). Structural information derived from the 3D model and breakdown of computed TMPKmt–THMDs interaction energies up to individual active site residue contributions helped us to design new more potent TMPKmt inhibitors. We obtained a reasonable agreement between the free energies of TMPKmt–THMDs complexation (ΔΔGcom) and Kexpi values, which explained approximately 93% of the TMPKmt inhibition data (pKi = −0.1422 ΔΔGcom + 4.9199, R2 = 0.93). Similar agreement was established for the PH4 pharmacophore model (pKexpi = 1.0016 × pKprei + 0.0077, R2 = 0.95). Comparative analysis of the active site residue contributions directed substitutions to various positions of the naphtholactam or naphthosultam moeties and suggested their replacement with phthalimido or isoindolinone or indanone rings, which led to a predicted increase of the inhibitory potency. The predicted Kprei for the best inhibitor candidate reached the picomolar range for aliphatic acyclic nucleoside analogs and for benzyl pyrimidine-like analogs. This computational approach, which combines molecular modelling, pharmacophore generation and analysis of TMPKmt–THMDs interaction energies resulted in a set of proposed TMPKmt inhibitors. It can thus direct medicinal chemists in their search for new antituberculotic agents.
1. Introduction
At this moment, we can be quite sure that the millennium development goal to halve the tuberculosis (TB) mortality relative to the 1990 level by 2015,1 will not be met. On the contrary, the number of countries reporting incidence of cases of multidrug-resistant (MDR) and extensively drug-resistant (XDR) tuberculosis, which are resistant to almost all fluoroquinolones plus the injectable antituberculotics, kanamycin, amikacin or capreomycin, is growing.2 Therefore, it becomes crucial to analyse why the TB burden increases despite treatment and past and current vaccination efforts.
According to WHO, Sub-Saharan Africa has the highest rates of TB driven primarily by the HIV co-infection, while approximately 2 billion individuals worldwide suffer from a latent mycobacterium infection and are at risk of developing active tuberculosis.3
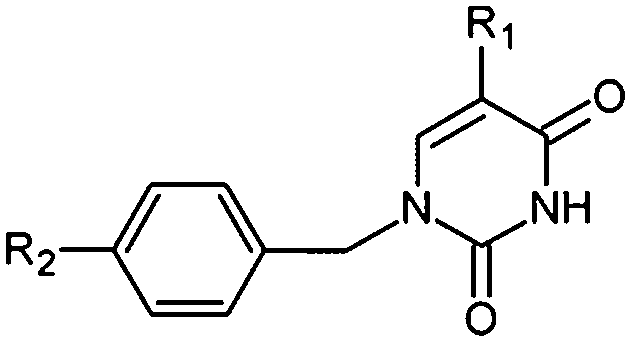
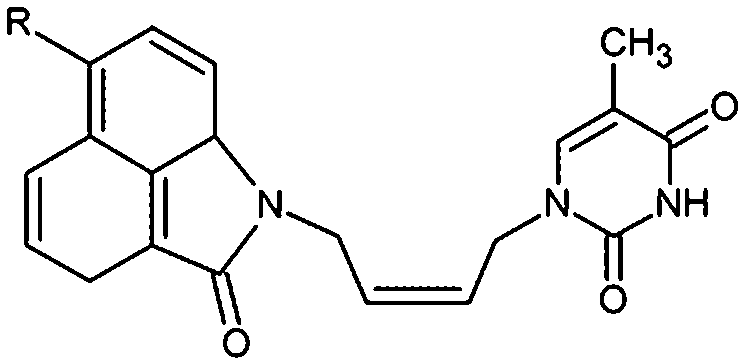
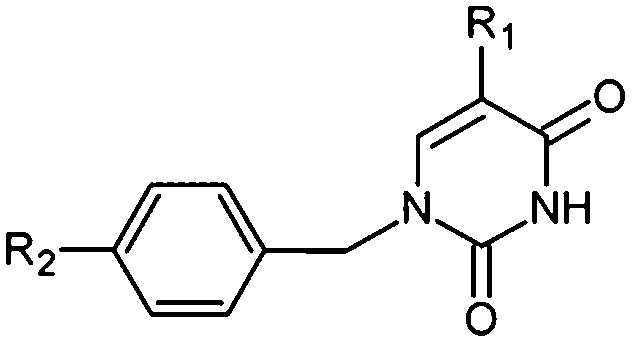
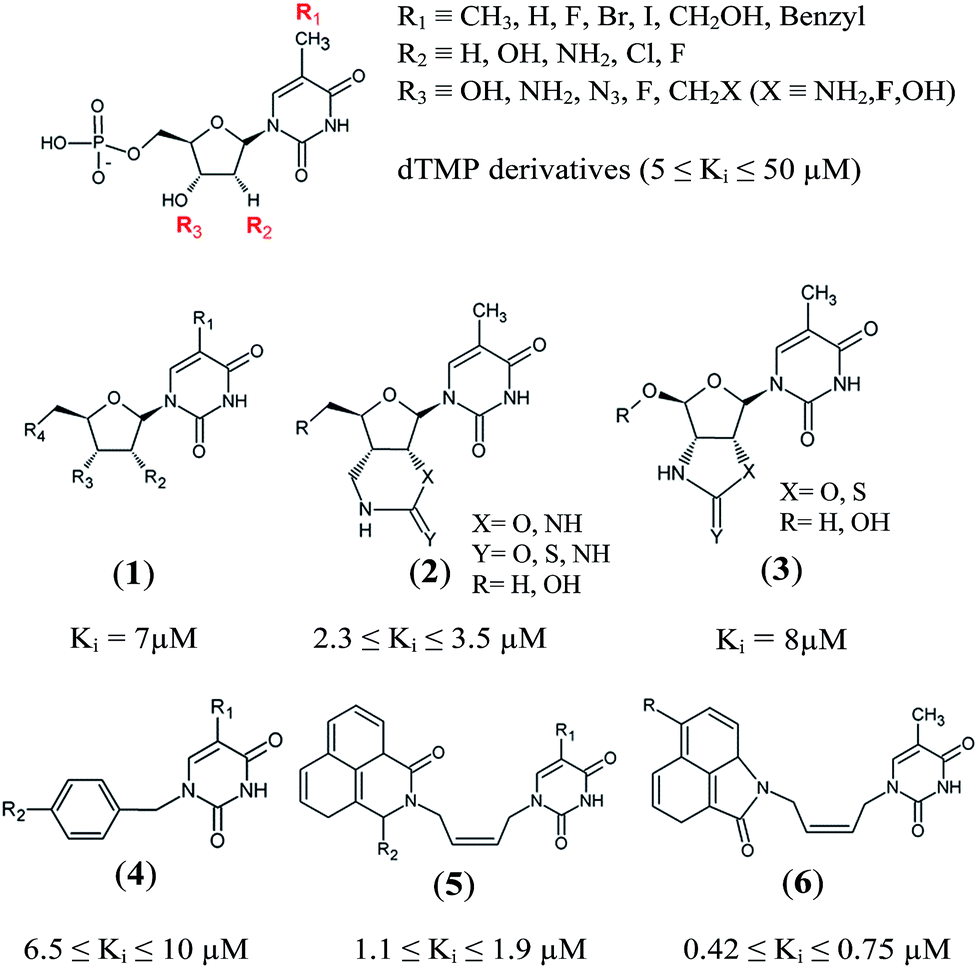
Thymidine monophosphate kinase (TMPKmt) was recognized as a validated pharmacological target (TMPKmt is an essential enzyme for the synthesis of thymidine diphosphate from deoxythymidine monophosphate (dTMP) that is needed for DNA synthesis and replication). TMPKmt inhibitors block the replication of the mycobacterium.4 So far, the most potent thymidine-derived inhibitors (THMD) reported in the literature exert their inhibitory activity in the low micromolar range (3.5–5 μM).5 More potent reported THMDs with inhibition constants within 0.27–0.75 μM contain aliphatic spacer linking the thymine base to the distal naphtholactam or naphthosultam moieties.6 Similarly, THMDs with aromatic spacers reached an inhibitory concentration range of 6.5–10 μM. Some THMD structures are shown in Fig. 1.
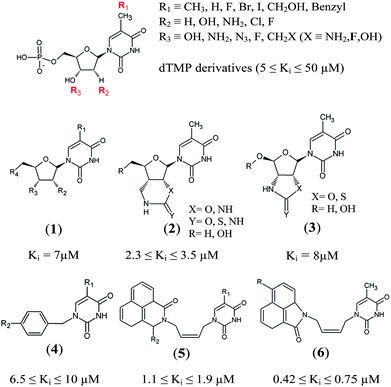 |
| | Fig. 1 Structures of known TMPKmt inhibitors. | |
The availability of X-rays crystal structures of the TMPKmt enzyme bound to its substrate as well as to several THMDs opened the gate to structure-based design of new antituberculotic agents sharing similar mode of action.7
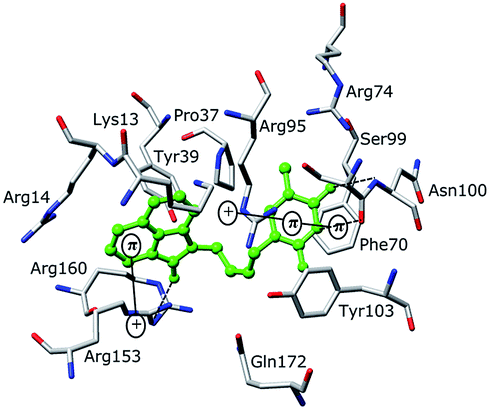
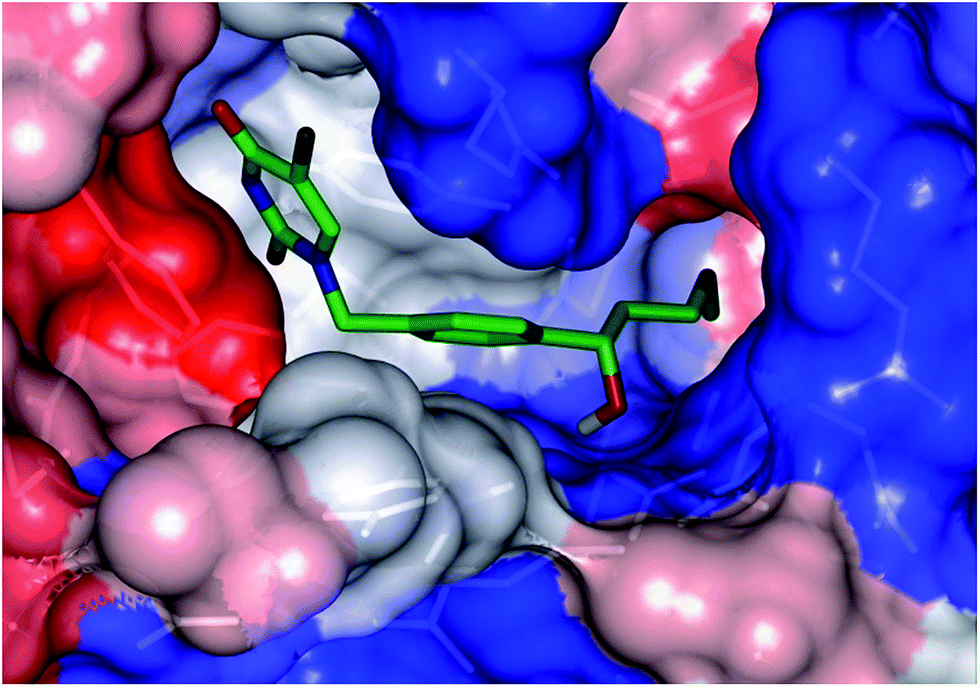
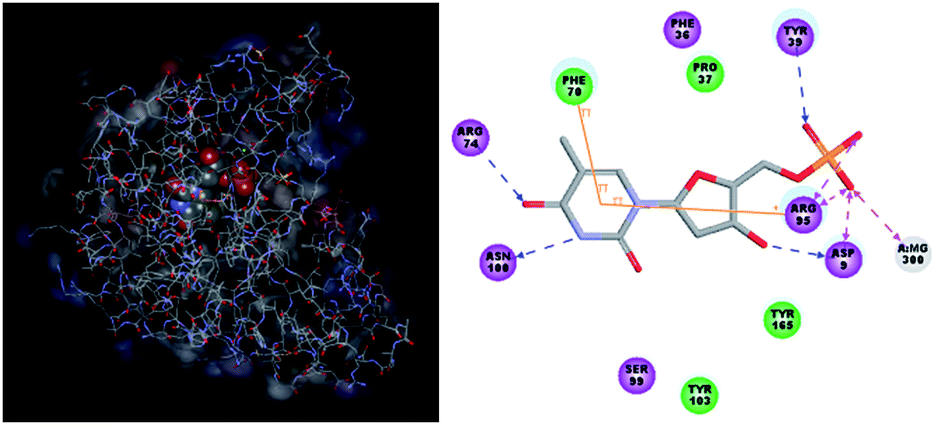
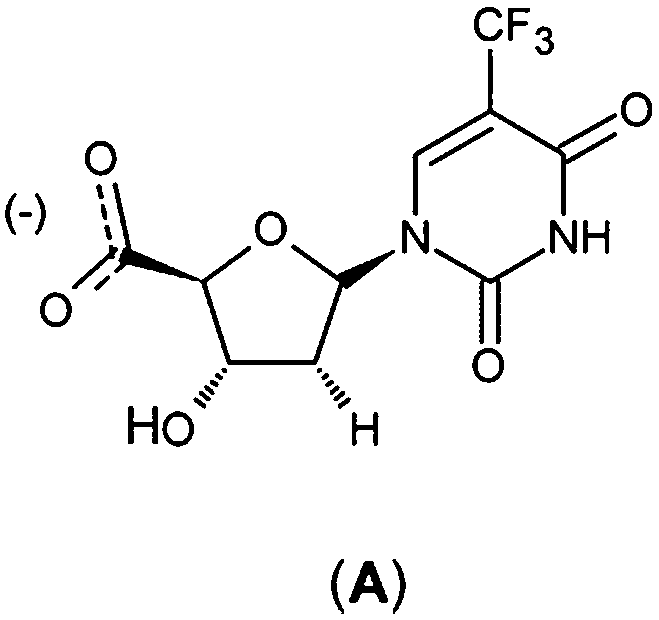
Structure-based design of TMPKmt inhibitors can start with assessment of enzyme–ligand interaction maps (Fig. 2), which can be extracted from high resolution crystal structures of TMPKmt–dTMP complexes, such as 1GSI8 and 1G3U.9–11 The maps show the pyrimidine ring of the thymine in a π–π stacking interaction with Phe70 and a hydrogen bond with Arg74 keeping the orientation of the pyrimidine ring, while the hydroxyl group of the ribose ring is hydrogen bonded to Asp9. The phosphate group is kept in its position by electrostatic interactions with the Mg2+ ion and by hydrogen bonding to Arg95. On the basis of this structural information dTMP analogs were proposed with the phosphate group replaced by anionic isosteres and bearing bromine substitution on the pyrimidine ring intended to improve the ADME profile of the compounds5,12 (Fig. 1, molecules (1–6)). The dTMP analogs with alkene linkers replacing the ribose moiety and dibenzoindolone or dibenzoquinolone rings exploring the edge to face interaction between the naphtyl group and the selective Tyr39 residue (5) and (6) represent submicromolar inhibitors of the TMPKmt.6 In a recent computational inhibitor design study a dTMP analogue with a carboxylate group in the 5′-position of the ribose ring was predicted to shift the inhibitory potency up to the nanomolar concentration range (Kprei = 0.155 nM; Fig. 3, inhibitor A).11
 |
| | Fig. 2 Interaction map of dTMP with active site residues (right) and location of the active site (CPK representation of dTMP) in the 3D model of TMPKmt (left). | |
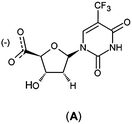 |
| | Fig. 3 Chemical structure of proposed nanomolar inhibitor A.11 | |
In this work we have extended the structure-based design of TMPKmt inhibitors and propose analogs that contain aromatic moieties attached by aliphatic or aromatic spacers to the thymine, which interact with the side chains of residues Tyr103, Tyr165 and Tyr39. A Hansch-type QSAR model of inhibitor–enzyme interaction was built for a training set of 20 known THMDs, which correlates computed Gibbs free energy of the enzyme–inhibitor complex formation with the experimental inhibition constants.6,12,13 In addition a 3D-QSAR model was used to prepare a four-feature pharmacophore (PH4) of the THMDs. The predictive power of the QSAR model of inhibitor–enzyme binding was cross-checked with the PH4 3D-QSAR pharmacophore model. This was used to screen a set of modelled thymidine analogs for potent TMPKmt inhibitors. The virtual hits identified by the complexation QSAR model reached predicted activities within subnanomolar concentration range.
2. Methods
2.1 Training and validation sets
The training and validation sets of thymine-like inhibitors of TMPKmt used in this study were taken from the literature.6,12,13 The inhibitory potencies of these derivatives cover sufficiently broad range of activity to allow a reliable QSAR model to be built (0.27μM ≤ Kexpi ≤ 202 μM).
2.2 Model building
Molecular models of the enzyme–inhibitor complexes (E:I), free TMPKmt (E) and inhibitors (I) were prepared from the high-resolution crystal structure (1.95 Å) of a reference complex containing the deoxythymidine monophosphate (dTMP) bound to TMPKmt8 (Protein Data Bank14 entry code 1G3U) using Discovery Studio 2.5 molecular modelling program.15
The structures of E and E:I complexes were considered to be at the pH of 7 with neutral N- and C-terminal residues and all protonizable and ionizable residues charged. No crystallographic water molecules were included into the model. The inhibitors were built into the reference structure by in situ replacing of the derivatized groups of the dTMP moiety (molecular scaffold). An exhaustive conformational search over all rotatable bonds of the replacing function group coupled with a careful gradual energy-minimization of the modified inhibitor and active site residues of the TMPKmt located in the vicinity of the inhibitor (within 5 Å distance), was employed to identify low-energy bound conformations of the modified inhibitor. The resulting low-energy structures of the E:I complexes were then carefully refined by minimization of the whole complex. This procedure has been successfully used for model building of viral, bacterial and protozoal enzyme–inhibitor complexes and design of peptidomimetic, hydroxynaphthoic and thymidine inhibitors.11,16–20
2.3 Molecular mechanics
Modelling of inhibitors, TMPKmt and complexes was carried out in all-atom representation using atomic and charge parameters of the CHARMm force field.15 A dielectric constant of 4 was used for all molecular mechanics (MM) calculations in order to take into account the dielectric shielding effect in proteins. Minimizations of the E:I complexes, free E and I were carried out by relaxing the structures gradually, starting with the added hydrogen atoms, continued with inhibitor heavy atoms, followed by residue side chains and concluded with protein backbone relaxation. In all the geometry optimizations, a sufficient number of steepest descent and conjugate gradient iterative cycles were used with the convergence criterion for the average gradient set to 0.01 kcal mol−1 Å−1.
2.4 Conformational search
Free inhibitor conformations were derived from their bound conformations in the E:I complexes by gradual relaxation to the nearest local energy minimum. Then a Monte Carlo search (with an upper limit of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations) for low-energy conformations over all rotatable bonds except those in the rings was carried out using Discovery Studio.15 Two hundred unique conformations were generated for each inhibitor by randomly varying torsion angles of the last accepted conformer by ±15° at 5000 K followed by subsequent energy minimization. During the minimization a dielectric constant ε = 80 was used to account approximately for the dielectric screening effect of hydration upon the generated conformers. The conformer with the lowest total energy was selected and re-minimized at ε = 4.
000 iterations) for low-energy conformations over all rotatable bonds except those in the rings was carried out using Discovery Studio.15 Two hundred unique conformations were generated for each inhibitor by randomly varying torsion angles of the last accepted conformer by ±15° at 5000 K followed by subsequent energy minimization. During the minimization a dielectric constant ε = 80 was used to account approximately for the dielectric screening effect of hydration upon the generated conformers. The conformer with the lowest total energy was selected and re-minimized at ε = 4.
2.5 Solvation Gibbs free energies
The electrostatic component of solvation Gibbs free energy that includes also the effects of ionic strength via solving nonlinear Poisson–Boltzmann equation21,22 was computed by the DelPhi module in Discovery Studio.15 The program treats the solvent as a continuous medium of high dielectric constant (εo = 80) and the solute as a cavity of low dielectric (εi = 4) with boundaries linked to the solute's molecular surface, which encloses the solute's atomic charges. The program uses a finite difference method to numerically solve for the molecular electrostatic potential and reaction field around the solute. DelPhi calculations were carried out on a (235 × 235 × 235) cubic lattice grid for the E:I complexes and free E and (65 × 65 × 65) grid for the free I with full coulombic boundary conditions. Two subsequent focusing steps led in both cases to a similar final resolution of about 0.3 Å per grid unit at 70% filling of the grid by the solute. Physiological ionic strength of 0.145 mol dm−3, atomic partial charges and radii defined in the CHARMm parameter set15 and a probe sphere radius of 1.4 Å were used. The electrostatic component of the solvation Gibbs free energy was calculated as the reaction field energy.21,23–25
2.6 Calculation of binding affinity
Inhibition constant (Ki) of a reversible inhibitor I is related to the standard Gibbs free energy (GFE) change of the formation of E:I complex (ΔGcom) in a solvent. The Ki value can thus be predicted from the complexation GFE as ΔGcom = −RTlnKi assuming the following equilibrium:| | |
{E}aq + {I}aq ↔ {E:I}aq
| (1) |
where { }aq indicates solvated species. The standard GFE change of reaction (1) can be derived by molecular simulations of the complex and the free reactants:| | |
ΔGcom = G{E:I} − G{E} − G{I}
| (2) |
In this work we approximate the exact values of standard GFE for larger systems such as enzyme–inhibitor complexes by the expression:18,19
| | |
G{E:I} ≈ EMM{E:I} + RT − TStrv{E:I} + Gsol{E:I}
| (3) |
where
EMM{E:I} stands for the molecular mechanics total energy of the complex (including bonding and non-bonding contributions),
Gsol{E:I} is the solvation GFE and TS
trv{E:I} is the entropic term:
| | |
TStrv{E:I} = TStran{E:I} + TSrot{E:I} + TSvib{E:I}
| (4) |
composed of a sum of contributions arising from translational, rotational and vibrational motions of E:I. Assuming that the tran and rot terms for the complex E:I and free enzyme E are approximately equal, we obtain:
| | |
ΔGcom ≈ [EMM{E:I} − EMM{E} − EMM{I}] + [Gsol{E:I} − Gsol{E} − Gsol{I}] + TStran{I} + TSrot{I} − [TSvib{E:I} − TSvib{E} − TSvib{I}] = ΔHMM + TStran{I} + TSrot{I} − ΔTSvib + ΔGsol
| (5) |
where TS
tran{I} and TS
rot{I} describe the translational and rotational entropy terms of the free inhibitor and ΔTS
vib represents a simplified vibrational entropy change upon the complex formation: ΔTS
vib = TS
vib{I}
E − TS
vib{I}.
26,27 Comparison between different inhibitors was done
via relative changes in the complexation GFE with respect to a reference inhibitor, I
ref, assuming ideal gas behaviour for the rotational and translational motions of the inhibitors:
| | |
ΔΔGcom = ΔGcom(I) − ΔGcom(Iref) = ΔΔHMM − ΔΔTSvib + ΔΔGsol
| (6) |
The binding energy calculation protocol in Discovery Studio 2.5.5 (the latest version of DS 2.5) can now compute the loss of conformational entropy of a bound ligand.28 This term is denoted ΔΔTS in Tables 2 and 5. The use of the entropic contribution leads to a more accurate evaluation of the relative binding affinity.
The evaluation of relative changes is preferable as it is expected to lead to partial cancellation of errors caused by the approximate nature of the molecular mechanics method as well as solvent and entropic effects description.
2.7 Interaction energy
To calculate the MM interaction energy (Eint) between enzyme residues and the inhibitor, a protocol available in Discovery Studio 2.5 (ref. 15) that computes the non-bonded interactions (van der Waals and electrostatic terms) between defined sets of atoms, was used. The calculations were performed using CHARMm force field15 with a dielectric constant of 4. The breakdown of Eint into active site residue contributions (presented in % of total Eint of TMPKmt–THMD) reveals the significance of individual interactions and allows a comparative analysis, which leads to identification of affinity enhancing and unfavourable THMD substitutions.
2.8 Pharmacophore generation
Pharmacophore modelling assumes that a set of structural features in a molecule is recognized by the receptor and is responsible for the molecule's biological activity. Bound conformations of inhibitors taken from the models of E:I complexes were used for building of 3D-QSAR pharmacophore by means of Catalyst HypoGen algorithm29 implemented in Discovery Studio 2.5.15 The top scoring pharmacophore hypothesis was built up in three stages (constructive, subtractive and optimization step) from the set of the most active inhibitors. Inactive molecules served for definition of the excluded volume. The maximum number of five features allowed by the HypoGen algorithm was selected based on the THMD scaffold and substituents during the pharmacophore generation, namely: hydrophobic aromatic (HYdAr), hydrophobic aliphatic (HYd), hydrogen-bond donor, (HBD), hydrogen-bond acceptor (HBA) and ring aromatic (Ar). Adjustable parameters of the protocol were kept at their default values except the uncertainty on the activity, which was set to 1.25 instead of 3. This parameter choice was intended to bring the uncertainty interval of experimental activity from wide span 〈Ki/3, 3 × Ki〉 to a relatively narrow one 〈4 × Ki/5, 5 × Ki/4〉 taking thus into account the accuracy and homogeneity of the measured inhibitory activities which are coming from the same laboratory. During generation of 10 pharmacophores the number of missing features was set to 0. The best pharmacophore model was selected.
2.9 ADME-related properties
Pharmacokinetics properties such as octanol–water partitioning coefficient, aqueous solubility, blood–brain partition coefficient, Caco-2 cell permeability, serum protein binding, number of likely metabolic reactions, and another eighteen descriptors related to adsorption, distribution, metabolism and excretion (ADME properties) of the inhibitors were computed by the QikProp program30 based on the method of Jorgensen.31–33 In this approach, experimental results of more than 710 compounds including about 500 drugs and related heterocycles were used to produce regression equations correlating experimental and computed descriptors resulting in an accurate prediction of molecule's pharmacokinetic properties. Drug likeness (#stars) − the number of property descriptors that fall outside the range of values determined for 95% of known drugs out of 24 selected descriptors computed by the QikProp,30 was used as an additional ADME-related compound selection criterion – the drug likeness.
3. Results and discussion
A training set of 20 THMDs (Table 1) and validation set of 3 THMVs (Table 1) were selected from 3 series of TMPKmt inhibitors with experimentally determined activities studied in the same laboratory.6,12,13 Their experimental inhibition constants Kexpi cover a concentration range sufficiently large (0.27–202 μM) to serve well for building of a useful QSAR model of TMPKmt inhibition.
Table 1 Training and validation sets of THMDs for the QSAR model
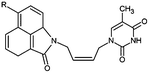
|
| Inhibitor |
R |
Ki (μM) |
| THMD5 |
H |
0.42 |
| THMD6 |
NO2 |
0.75 |
| THMD7 |
SO2 |
0.27 |
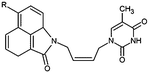
|
| Inhibitor |
R |
Ki (μM) |
| THMV1 |
NCHCOCH3 |
6 |
| THMV3 |
NH2 |
2.4 |
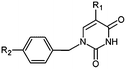
|
| Inhibitor |
R1 |
R2 |
Ki (μM) |
| THMV2 |
Br |
(CH2)3CONH2 |
39 |
3.1 QSAR model
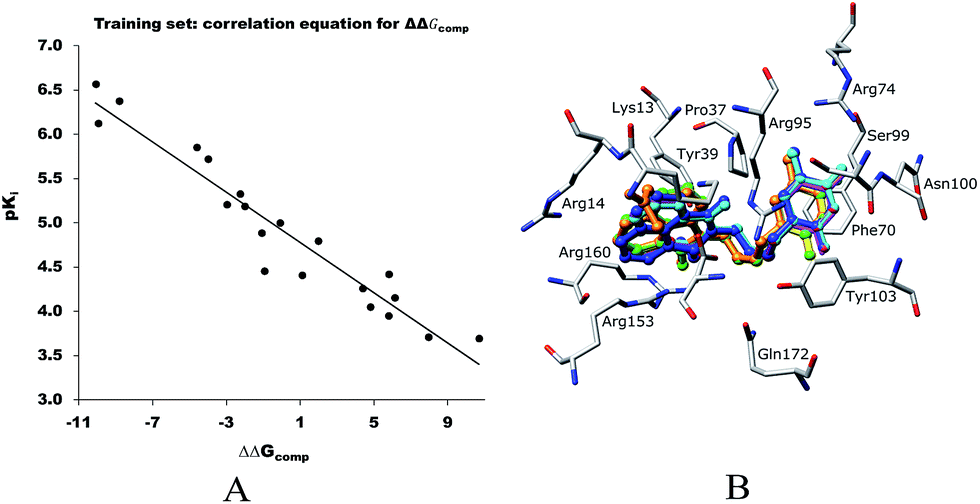
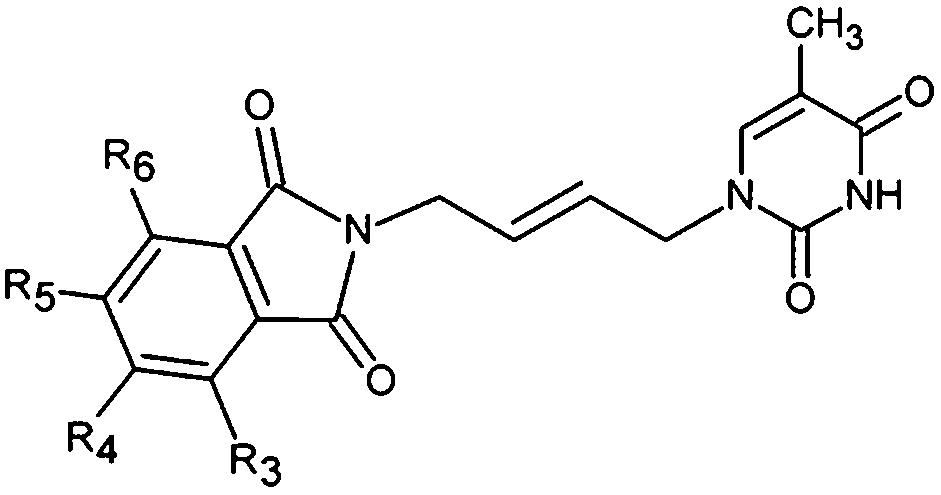
The relative Gibbs free energy of the E:I complex formation, eqn (6), was computed for the complexes prepared by in situ modification of the template inhibitor dTMP in the binding site of TMPKmt as described in the Methods section. Table 2 lists the ΔΔGcom of E:I binding and its components, eqn (6). Since it is computed in an approximate way, the relevance of the binding model is evaluated through a correlation with the experimental activity data (Kexpi) by a linear regression. The statistical data of the obtained regression equation are presented in Fig. 4A and listed in Table 3. Relatively high values of the regression coefficient and Fischer F-test indicate that there is a strong relationship between the binding model and the observed inhibitory potencies of the THMDs. The ratio of predicted and observed inhibition constants (Kprei/Kexpi) for the validation set of THMVs (not included into the training set) is close to one and documents considerable predictive power of the QSAR model. Therefore, the regression equation and computed ΔΔGcom quantities can be used to predict TMPKmt inhibition constants Kprei of novel THMD analogs, provided that the analogs share the same binding mode with the training set compounds.
Table 2 Complexation Gibbs free energy (binding affinity) and its components for the training set of TMPKmt inhibitors THMD1–20 and validation set inhibitors THMV1–3
| Training seta |
Mwb [g mol−1] |
ΔΔHMMc [kcal mol−1] |
ΔΔGsold [kcal mol−1] |
ΔΔTSe [kcal mol−1] |
ΔΔGcomf [kcal mol−1] |
Kexpig [μM] |
| For the chemical structures of the training and validation sets of inhibitors see Table 1. Mw is the molecular mass of the inhibitor. ΔΔHMM is the relative enthalpic contribution to the Gibbs free energy change related to the TMPKmt:THMD complex formation derived by molecular mechanics (MM): ΔΔHMM ≅ [EMM{TMPKmt:THMDx} − EMM{THMDx}] − [EMM{TMPKmt:THMD0} − EMM{THMD0}], THMD0 – is the reference inhibitor molecule (1) with R1 = Me, R2 = H, R3 = OH and R4 = N3. ΔΔGsolv is the relative solvation Gibbs free energy contribution to the Gibbs free energy change related to TMPKmt:THMD complex formation: ΔΔGsol = [Gsol{TMPKmt:THMDx} − Gsol{THMDx}] − [Gsol{TMPKmt:THMD0} − Gsol{THMD0}]. ΔΔTS is the relative entropic contribution of the inhibitor to the Gibbs free energy related to protease–inhibitor complex formation: ΔΔTS = [TS{TMPKmt:THMDx} − TS{THMDx}] − [TS{TMPKmt:THMD0} − TS{THMD0}]. ΔΔGcom is the relative Gibbs free energy change related to the enzyme–inhibitor complex formation: ΔΔGcom ≅ ΔΔHMM + ΔΔGsol − ΔΔTSvib. Kexpi is the experimental TMPKmt inhibition constant taken from ref. 6, 12 and 13. Ratio of predicted and experimental inhibition constants Kprei/Kexpi. Kprei was derived from pKprei = −log10(Kprei) which was predicted from computed ΔΔGcom using the regression equation shown in Table 3. |
| THMD1 |
375 |
−1.40 |
−1.67 |
0.93 |
−4 |
1.9 |
| THMD2 |
439 |
−4.32 |
3.02 |
0.96 |
−2.26 |
4.7 |
| THMD3 |
377 |
−6.36 |
4.42 |
1.04 |
−2.98 |
6.2 |
| THMD4 |
391 |
−4.25 |
0.49 |
0.85 |
−4.61 |
1.4 |
| THMD5 |
361 |
−18.42 |
10.36 |
0.75 |
−8.81 |
0.42 |
| THMD6 |
392 |
−24.14 |
15.30 |
1.11 |
−9.95 |
0.75 |
| THMD7 |
383 |
−20.63 |
11.47 |
0.94 |
−10.09 |
0.27 |
| THMD8 |
287 |
1.69 |
3.47 |
0.36 |
4.8 |
89 |
| THMD9 |
288 |
1.46 |
3.29 |
0.37 |
4.38 |
55 |
| THMD10 |
285 |
2.96 |
5.35 |
0.37 |
7.94 |
195 |
| THMD11 |
301 |
1.36 |
4.97 |
0.54 |
5.79 |
112 |
| THMD12 |
284 |
3.33 |
3.22 |
0.43 |
6.12 |
70 |
| THMD13 |
366 |
−18.90 |
19.80 |
1 |
−0.1 |
10 |
| THMD14 |
294 |
6.36 |
0.55 |
1.11 |
5.8 |
38 |
| THMD15 |
288 |
3.04 |
8.09 |
0.44 |
10.69 |
202 |
| THMD16 |
323 |
−16.93 |
15.66 |
0.73 |
−2.01 |
6.5 |
| THMD17 |
302 |
−9.20 |
8.65 |
0.55 |
−1.1 |
13 |
| THMD18 |
322 |
−7.83 |
9.66 |
0.73 |
1.1 |
39 |
| THMD19 |
337 |
−5.59 |
8.44 |
0.88 |
1.97 |
16 |
| THMD20 |
380 |
−19.70 |
19.87 |
1.11 |
−0.95 |
35 |
| Validation set |
Mw [g mol−1] |
ΔΔHMM [kcal mol−1] |
ΔΔGsol [kcal mol−1] |
ΔΔTS [kcal mol−1] |
ΔΔGcom [kcal mol−1] |
Kprei/Kexpih |
| THMV1 |
404 |
−18.51 |
17.30 |
1.21 |
−2.42 |
0.907 |
| THMV2 |
366 |
−9.04 |
13.08 |
1.00 |
3.05 |
0.837 |
| THMV3 |
362 |
−17.38 |
13.50 |
0.88 |
−4.76 |
1.054 |
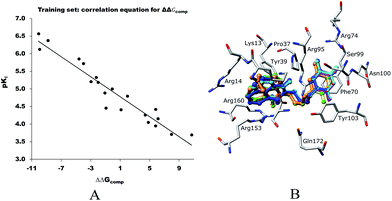 |
| | Fig. 4 A. Plot of the correlation equation between pKi and relative complexation Gibbs free energies ΔΔGcom [kcal mol−1] of the training set. B. Superimposition of optimized bound conformations of highly active training set compounds. | |
Table 3 Regression analysis of computed binding affinities ΔΔGcom and experimental inhibition constants pKexpi = −log10(Kexpi) of THMDs towards TMPKmt
| Statistical data of regression |
| pKi = −0.1422ΔΔGcom + 4.9199 |
| Number of compounds n |
20 |
| Squared correlation coefficient of regression R2 |
0.93 |
| Cross-validated squared correlation coefficient RxV2 |
0.92 |
| Standard error of regression σ |
0.248 |
| Statistical significance of regression, Fischer F-test |
218.9 |
| Level of statistical significance α |
>95% |
| Range of activity of Kexpi [μM] |
0.27–202 |
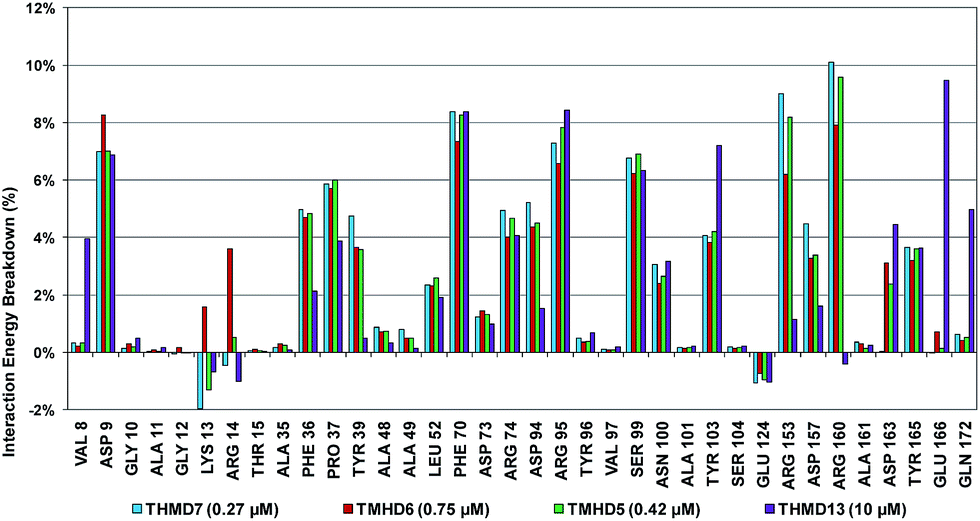
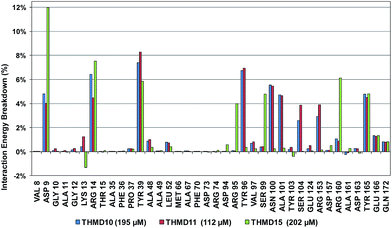
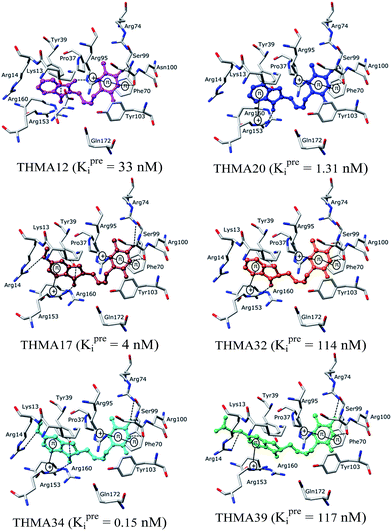
To identify chemical modifications of THMDs leading to new inhibitor structures with high predicted binding to TMPKmt, we have analyzed contributions of individual residues of the enzyme binding pocket to the total computed enzyme–inhibitor interaction energy (Eint). In this way we have noticed a significant contribution to the Eint from residues Phe70, Arg95, Ser99 with a strong variation of the contributions of Arg153 and Arg160 for the highly active THMDs (Fig. 7) and less active inhibitors (Fig. 8).
3.2 Binding mode of inhibitors
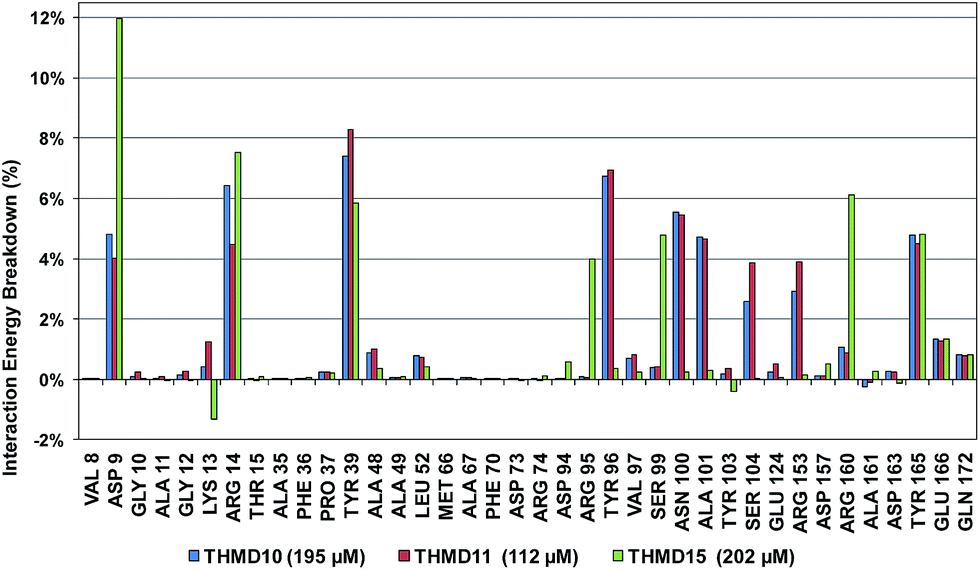
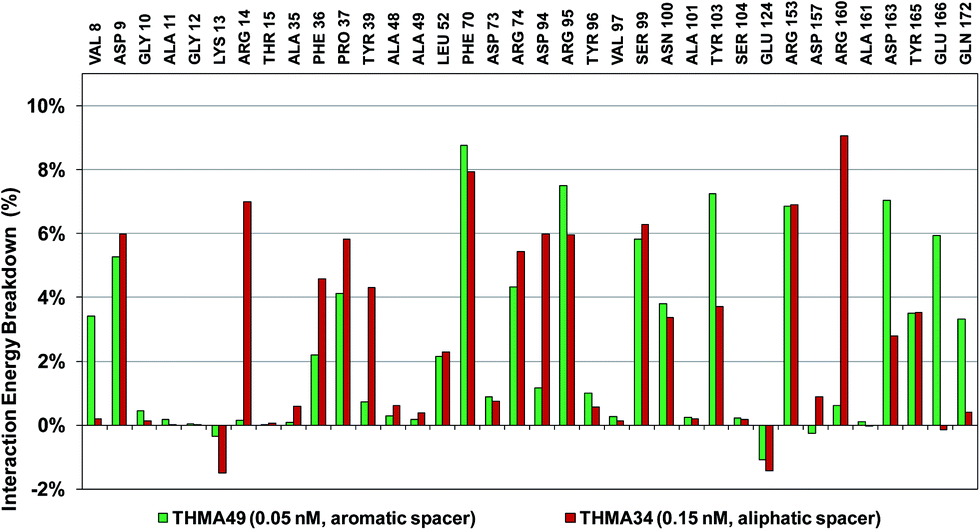
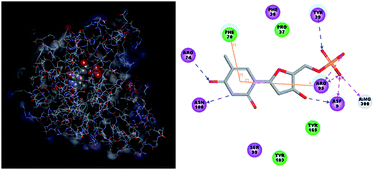
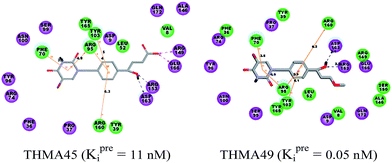
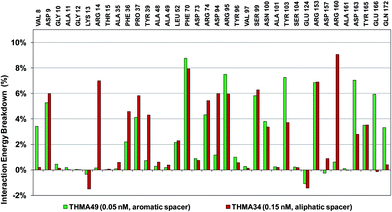
The binding mode of THMDs in the active site of TMPKmt present in the crystal structures of TMPKmt–dTMP complexes7–9 which was used in the complexation model of inhibitors THMD1–THMD20 is illustrated in Fig. 5 and 6. In this model the main electrostatic and hydrogen bonding interactions of the phosphate moiety of dTMP with residues Arg95 (and Tyr39) are preserved (Fig. 7). The stacking interaction of the pyrimidine ring with Phe70 is kept along with the cation–aromatic π interaction with Arg95 and Arg153. This last interaction is probably the main driving force of the increase of affinity of the submicromolar inhibitors THMD5–THMD7 with the TMPKmt. A comparison of the interaction patterns of Fig. 7 and 8 highlights the main stabilizing contributions of individual residues to the Eint (in % of the total Eint of TMPKmt:THMD) for the highly active THMDs (hTHMDs) namely THMD5 (0.42 μM), THMD6 (0.75 μM), THMD7 (0.27 μM) and THMD13 (10 μM) (Fig. 7) and for less active ones (lTHMDs) THMD10 (195 μM), THMD11 (112 μM) and THMD15 (202 μM) (Fig. 8). The essential contributions of residues to Eint expressed in % of total Eint for hTHMDs vs. lTHMDs for Phe36 (5% vs. 0%), Pro37 (6% vs. 0%), Phe70 (8% vs. 0%), Arg74 (5% vs. 0%), Asp94 (5% vs. 1%), Arg95 (8% vs. 4%), Arg153 (8% vs. 3%) and Arg160 (10% vs. 4%) known to be significant to the binding of the potent analogs globally are missing for the less active THMDs. Oppositely Tyr96 (0% vs. 7%) contribution is missing for hTHMDs can be used for the design of new potent analogs. A refined analysis of hTHMDs (Fig. 7) reveals the essential contribution specific to aromatic spacer THMDs (arTHMDs) and missing for aliphatic spacer THMDs (alTHMDs):Glu166 (9% for THMD13 vs. 0% for THMD5–7), Gln172 (5% vs. 1%) and Val8 (4% vs. 0%). Improvement in the contribution of these last three residues lead to more potent arTHMDs analogs exemplified by THMA49 and 50. The increased contribution of Tyr103 (7% for THMD13 vs. 4% for THMD5–7) is due to additional aromatic π–π interaction with the aromatic spacer as depicted in 2D in Fig. 11.
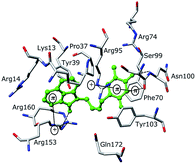 |
| | Fig. 5 Close-up of one of the most active training set inhibitor THMD5 (green color) at the active site of TMPKmt. Hydrogen bonds are shown in dashed black lines, other interactions are displayed as solid black lines. | |
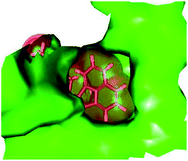 |
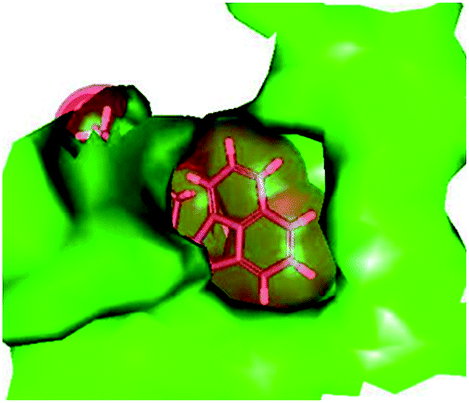
| | Fig. 6 Naphtholactam moiety in stick representation fitted into the binding pocket of the TMPKmt. Solid molecular surface of the active site is shown in green colour. | |
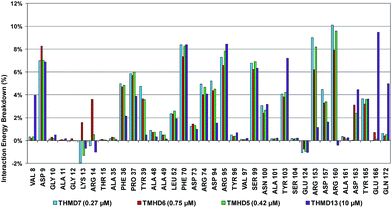 |
| | Fig. 7 Interaction energy breakdown (in % of the total Eint of TMPKmt:THMD) to residue contributions for highly active training set THMDs. | |
 |
| | Fig. 8 Interaction energy breakdown (in % of the total Eint of TMPKmt:THMD) to residue contributions for less active training set THMDs. | |
For alTHMDs Fig. 5 and 7 indicate that positions 6 and 7 of the naphtholactam are suitable for favourable interactions with Lys13 while positions 4 and 5 can be used for interaction with Arg14.
3.3 Pharmacophore model of inhibitory activity
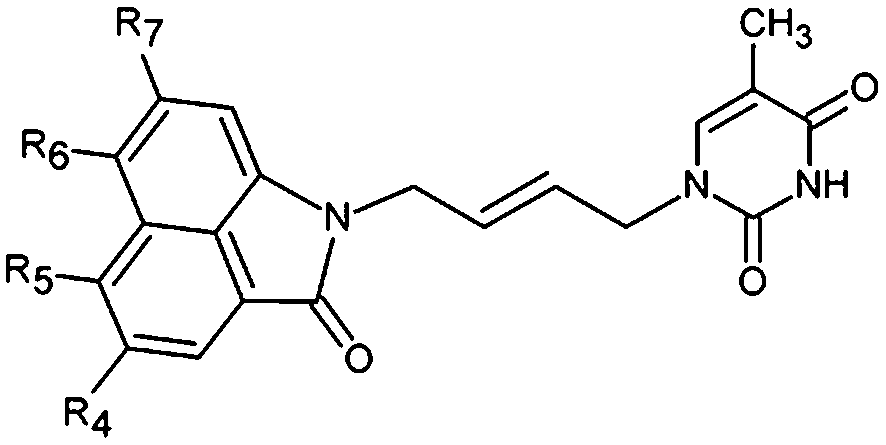
The 3D-QSAR PH4 pharmacophore generation process follows three main steps, the constructive, the subtractive and the optimization steps. The constructive phase of HypoGen auto-matically selected as leads the most active compounds for which 0.27 × 1.25 − Kexpi/1.25 > 0, namely 0.27 ≤ Kexpi ≤ 0.42 μM, THMD5 and THMD7 were used to generate all starting PH4 features and only those features were retained which matched both leads THMD5, THMD7. In the subtractive phase the inactive compounds with log10(Kexpi) − log10(0.27) > 3.5 were used to remove those pharmacophoric features that mapped more than 50% of these compounds, while pharmacophore representatives which contained all five features were retained. In the optimization phase, only the highest scoring PH4s based on their probability function cost which was calculated by a simulated annealing protocol, were retained. A total of 10 optimized hypotheses were kept all displaying four features. The costs ranged from 89.7 (Hypo1) to 190.5 (Hypo10). The relatively small gap between the highest and the lowest cost corresponds well with the homogeneity of the generated hypotheses and the consistency of the training set. For this PH4 the fixed cost (47.9) is lower than the null cost (810.3) by a difference Δ = 762.4. This difference is a major quality indicator of the PH4 predictability (it has to be noted that Δ > 70 corresponds to an excellent chance or a probability higher than 90% that the model represents a true correlation15). To be statistically significant the hypotheses have to be as close as possible to the fixed cost and as far as possible from the null cost. For the set of 10 hypotheses the difference was larger or equal to 762.4, which attests high quality of the pharmacophore model. The standard indicators as the root-mean-square deviations (RMSD) between the hypotheses range from 1.998 to 3.732 and the squared correlation coefficient (R2) falls to an interval from 0.97 to 0.91. The first PH4 hypothesis with the best cost, RMSD and R2 was retained for further analysis. The statistical data for the set of hypotheses (costs, RMSD, R) are listed in Table 4. The regression equation for Hypo1: pKexpi = 1.0016 × pKprei + 0.0077 (n = 20, R2 = 0.95, RxV2 = 0.94, F = 330.8, σ = 0.204, α > 95%) is plotted in Fig. 9. The figure shows also the geometry of the Hypo1 PH4 and the THMV3 inhibitor mapping to it. To check the consistency of the generated pharmacophore model we have calculated the ratio of predicted and observed activities (Kprei/Kexpi) for the validation set. The computed ratios were as follows: THMV1, 1.019; THMV2, 0.918; THMV3, 1.068; all of them lie relatively close to one, which documents a substantial predictive power of the regression for the best PH4 model. The randomization validation of the PH4 model was also carried out by the CatScramble algorithm in the Catalyst for 49 random runs corresponding to 98% confidence level. This procedure created 10 valid hypotheses for each run, however, none of them was as predictive as the Hypo10, the hypothesis with the highest cost is shown in Table 4. Thus there is a 98% probability that the best selected hypothesis Hypo1 represents a pharmacophore model for the biological activity of THMDs within the same level of predictive power as the complexation model, which relies on the 3D structures of the TMPKmt:THMDx complexes and computed Gibbs free energies of the enzyme–inhibitor binding ΔΔGcom.
Table 4 Output parameters of 10 generated PH4 pharmacophoric hypotheses after CatScramble validation procedure for TMPKmt inhibitors listing RMSD, total cost and correlation coefficient R
| Hypothesis |
RMSD |
R |
Total Cost |
| Hypo1 |
1.998 |
0.97 |
89.7 |
| Hypo2 |
2.733 |
0.95 |
123.9 |
| Hypo3 |
2.921 |
0.94 |
135.2 |
| Hypo4 |
3.038 |
0.94 |
141.4 |
| Hypo5 |
3.182 |
0.93 |
150.1 |
| Hypo6 |
3.701 |
0.91 |
188.1 |
| Hypo7 |
3.699 |
0.91 |
188.3 |
| Hypo8 |
3.703 |
0.91 |
188.8 |
| Hypo9 |
3.707 |
0.91 |
188.9 |
| Hypo10 |
3.732 |
0.91 |
190.5 |
| Fixed cost |
0.0 |
1.0 |
47.9 |
| Null cost |
8.802 |
0.0 |
810.3 |
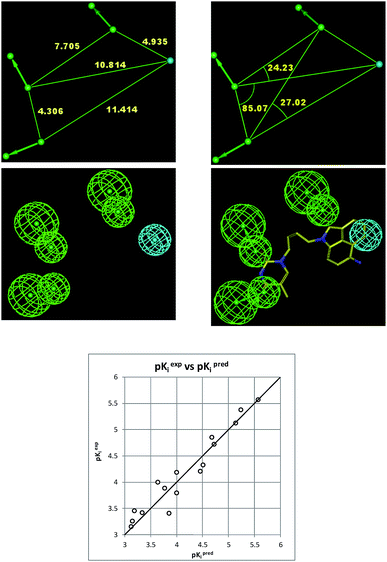 |
| | Fig. 9 Coordinates (top), features (middle left) and mapping (middle right) of the TMPKmt inhibition pharmacophore with THMV3 (yellow). The correlation plot experimental vs. predicted inhibitory activity is displayed at the bottom. | |
3.4 New inhibitors
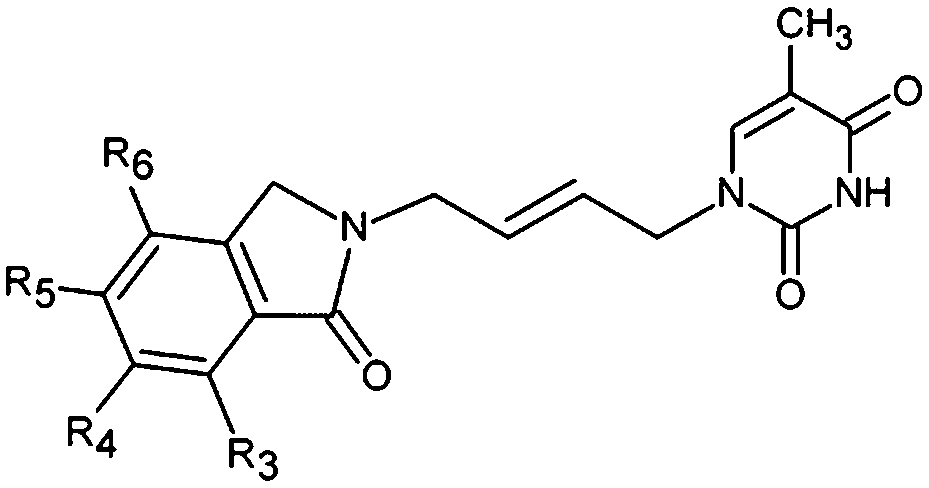
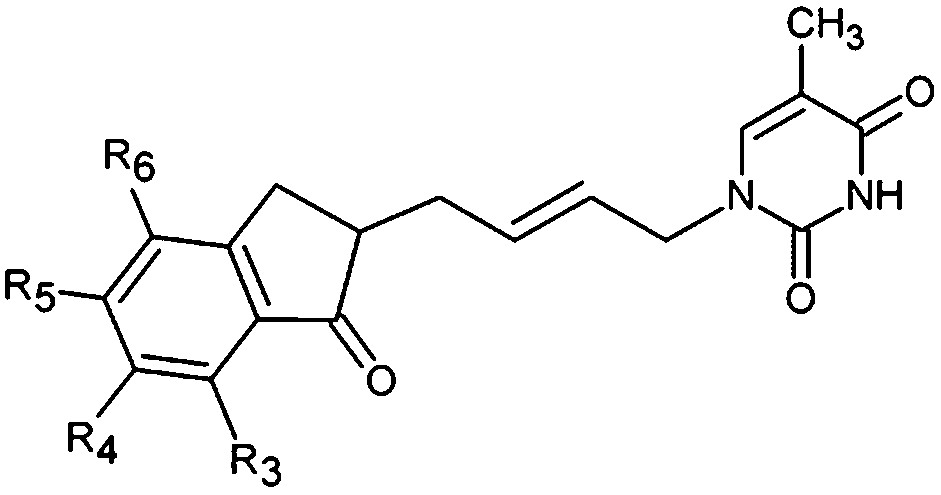
The design of new analogs in this work is based either on the favourable features of the naphtholactam, phthalimide, isoindoline and indanone moieties linked via 2-butene spacer to the thymine or the carboxylic, hydroxymethyl moiety via aromatic spacer, Table 5.5,6 We have noticed that certain substitutions in positions 4 to 7 of the naphtholactam ring and in positions of 3 to 6 of the related phthalimide, isoindoline and indanone moieties with a reduced size lead to elevated predicted binding affinities of the modelled analogs to the TMPKmt due to favourable attractive interactions of the substituents with the cationic residues Lys13 and Arg14, which are located in the vicinity of the substitution sites. This is the reason why some of the novel analogs (THMA1–50, Table 5) bearing negatively charged substituent in some of these positions were predicted to display inhibition potencies in the picomolar range. The Kprei were calculated from the regression equation of the QSAR model of TMPKmt inhibition by THMDs (Table 3). The substitutions and molecular scaffolds considered are listed for the five considered categories of the THMAs in Table 5 and are shown on Fig. 10–12.
Table 5 Designed THMD analogs (THMA1–50) and their predicted inhibitory activities (Kprei) against TMPKmt
|
| Inhibitor |
Structure |
ΔΔHMM |
ΔΔGsol |
ΔΔTS |
ΔΔGcom |
Kprei [nM] |
| R4 |
R5 |
R6 |
R7 |
| THMA1 |
H |
H |
OH |
H |
−24.72 |
10.19 |
0.90 |
−15.43 |
77 |
| THMA2 |
H |
H |
H |
OH |
−26.14 |
9.37 |
0.87 |
−17.64 |
37 |
| THMA3 |
H |
CH2OH |
H |
H |
−24.05 |
5.27 |
1.01 |
−19.8 |
18 |
| THMA4 |
H |
H |
CH2OH |
H |
−24.59 |
6.39 |
1.02 |
−19.23 |
22 |
| THMA5 |
H |
H |
H |
CH2OH |
−23.89 |
4.91 |
0.96 |
−19.93 |
18 |
| THMA6 |
H |
CH2OH |
CH2OH |
H |
−24.52 |
1.51 |
1.21 |
−24.22 |
4 |
| THMA7 |
H |
H |
SO2Cl |
H |
−32.57 |
11.41 |
1.45 |
−22.61 |
7 |
| THMA8 |
H |
H |
Cl |
H |
−21.50 |
3.57 |
1.05 |
−18.97 |
24 |
| THMA9 |
H |
H |
F |
H |
−21.36 |
3.61 |
0.91 |
−18.66 |
27 |
| THMA10 |
H |
H |
Br |
H |
−21.16 |
3.40 |
1.31 |
−19.07 |
23 |
| THMA11 |
H |
H |
Cl |
Cl |
−19.56 |
3.50 |
0.90 |
−15.43 |
77 |
|
| Inhibitor |
Structure |
ΔΔHMM |
ΔΔGsol |
ΔΔTS |
ΔΔGcom |
Kprei [nM] |
| R3 |
R4 |
R5 |
R6 |
| THMA12 |
H |
H |
H |
H |
−29.51 |
12.06 |
0.56 |
−18.02 |
33 |
| THMA13 |
H |
NHCOCH3 |
H |
H |
−27.33 |
11.10 |
1.05 |
−17.28 |
42 |
| THMA14 |
H |
NO2 |
H |
H |
−24.51 |
12.29 |
0.56 |
−12.78 |
183 |
| THMA15 |
H |
NH2 |
H |
H |
−25.82 |
8.09 |
0.71 |
−18.44 |
29 |
| THMA16 |
H |
COO(−) |
H |
H |
−31.87 |
6.95 |
0.99 |
−23.14 |
6 |
| THMA16′ |
H |
H |
COO(−) |
H |
−24.17 |
4.40 |
0.99 |
−20.76 |
13 |
| THMA17 |
H |
OCH3 |
H |
H |
−28.90 |
4.92 |
0.85 |
−24.82 |
4 |
| THMA18 |
OCH3 |
OCH3 |
H |
H |
−30.62 |
9.18 |
1.03 |
−22.48 |
8 |
| THMA19 |
OCH3 |
H |
OCH3 |
H |
−29.87 |
12.65 |
1.08 |
−18.3 |
30 |
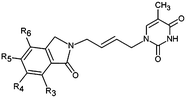
|
| Inhibitor |
Structure |
ΔΔHMM |
ΔΔGsol |
ΔΔTS |
ΔΔGcom |
Kprei [nM] |
| R3 |
R4 |
R5 |
R6 |
| THMA20 |
H |
H |
H |
H |
−29.06 |
1.70 |
0.52 |
−27.88 |
1.31 |
| THMA21 |
H |
H |
NO2 |
H |
−21.02 |
−1.97 |
0.96 |
−23.95 |
4.7 |
| THMA22 |
H |
H |
NH2 |
H |
−19.45 |
−1.95 |
0.68 |
−22.08 |
8.7 |
| THMA23 |
H |
H |
COO(−) |
H |
−27.06 |
2.11 |
0.95 |
−25.9 |
2.5 |
| THMA24 |
H |
COO(−) |
H |
H |
−27.15 |
8.12 |
0.91 |
−19.94 |
18 |
| THMA25 |
H |
H |
NHCOCH3 |
H |
−26.08 |
1.61 |
1.09 |
−25.55 |
2.80 |
| THMA26 |
CH3 |
H |
NHCOCH3 |
H |
−27.45 |
−1.83 |
0.99 |
−30.28 |
0.60 |
| THMA27 |
CH2CH3 |
H |
NHCOCH3 |
H |
−39.69 |
6.97 |
1.27 |
−33.99 |
0.18 |
| THMA28 |
H |
H |
OCH3 |
H |
−23.28 |
7.09 |
0.50 |
−16.7 |
51 |
| THMA29 |
H |
OCH3 |
OCH3 |
H |
−24.82 |
1.11 |
1.05 |
−24.75 |
4 |
| THMA30 |
CH3 |
OCH3 |
OCH3 |
H |
−23.00 |
0.86 |
1.13 |
−23.27 |
6 |
| THMA31 |
Cl |
Cl |
Cl |
Cl |
−20.18 |
−7.84 |
0.86 |
−28.88 |
0.94 |
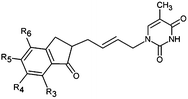
|
| Inhibitor |
Structure |
ΔΔHMM |
ΔΔGsol |
ΔΔTS |
ΔΔGcom |
Kprei [nM] |
| R3 |
R4 |
R5 |
R6 |
| THMA32 |
H |
H |
H |
H |
−18.84 |
5.18 |
0.57 |
−14.23 |
114 |
| THMA33 |
H |
H |
NH2 |
H |
−20.54 |
−2.13 |
0.96 |
−23.63 |
5 |
| THMA34 |
H |
H |
OCH3 |
H |
−24.37 |
−9.06 |
1.17 |
−34.6 |
0.15 |
| THMA35 |
H |
OCH3 |
OCH3 |
H |
−22.61 |
0.04 |
1.04 |
−23.61 |
5 |
| THMA36 |
NH2 |
H |
OCH3 |
H |
−19.31 |
−2.91 |
0.93 |
−23.15 |
6 |
| THMA37 |
COO(−) |
H |
OCH3 |
H |
−13.22 |
−7.17 |
1.12 |
−14.48 |
105 |
| THMA38 |
H |
H |
NHCOOH |
H |
−25.28 |
0.56 |
1.09 |
−25.81 |
2.6 |
| THMA39 |
H |
H |
OCH2CO2− |
H |
−32.82 |
19.95 |
1.29 |
−14.16 |
117 |
| THMA40 |
H |
H |
COOC2H5 |
H |
−26.75 |
2.86 |
1.22 |
−25.11 |
3 |
| THMA41 |
CH2CO2− |
H |
OCH3 |
H |
−19.89 |
9.74 |
1.25 |
−11.41 |
287 |
| THMA42 |
H |
H |
OCH3 |
OCH3 |
−26.37 |
2.16 |
0.98 |
−25.19 |
3 |
| THMA43 |
H |
H |
NHCOCH3 |
H |
−27.99 |
1.34 |
1.08 |
−27.73 |
1.37 |
| THMA44 |
H |
H |
COO(−) |
H |
−32.07 |
21.52 |
0.95 |
−11.5 |
278 |
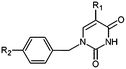
|
| Inhibitor |
R1 |
R2 |
ΔΔHMM |
ΔΔGsol |
ΔΔTS |
ΔΔGcom |
Kprei [nM] |
| THMA45 |
Br |
CHOH(CH2)2CO2− |
−50.51 |
30.22 |
1.07 |
−21.36 |
11 |
| THMA46 |
Br |
CHOH(CH2)2CO2CH3 |
−27.41 |
19.00 |
1.21 |
−9.63 |
514 |
| THMA47 |
Br |
(CHOH)2CH2CO2− |
−52.01 |
34.75 |
1.19 |
−18.45 |
29 |
| THMA48 |
CH3 |
CHOH(CH2)2CO2− |
−50.30 |
30.22 |
0.68 |
−20.76 |
13 |
| THMA49 |
Br |
CHOH(CH2)2OCH3 |
−46.22 |
8.83 |
0.96 |
−38.35 |
0.05 |
| THMA50 |
CH3 |
CHOH(CH2)2OCH3 |
−45.98 |
8.68 |
0.51 |
−37.81 |
0.06 |
 |
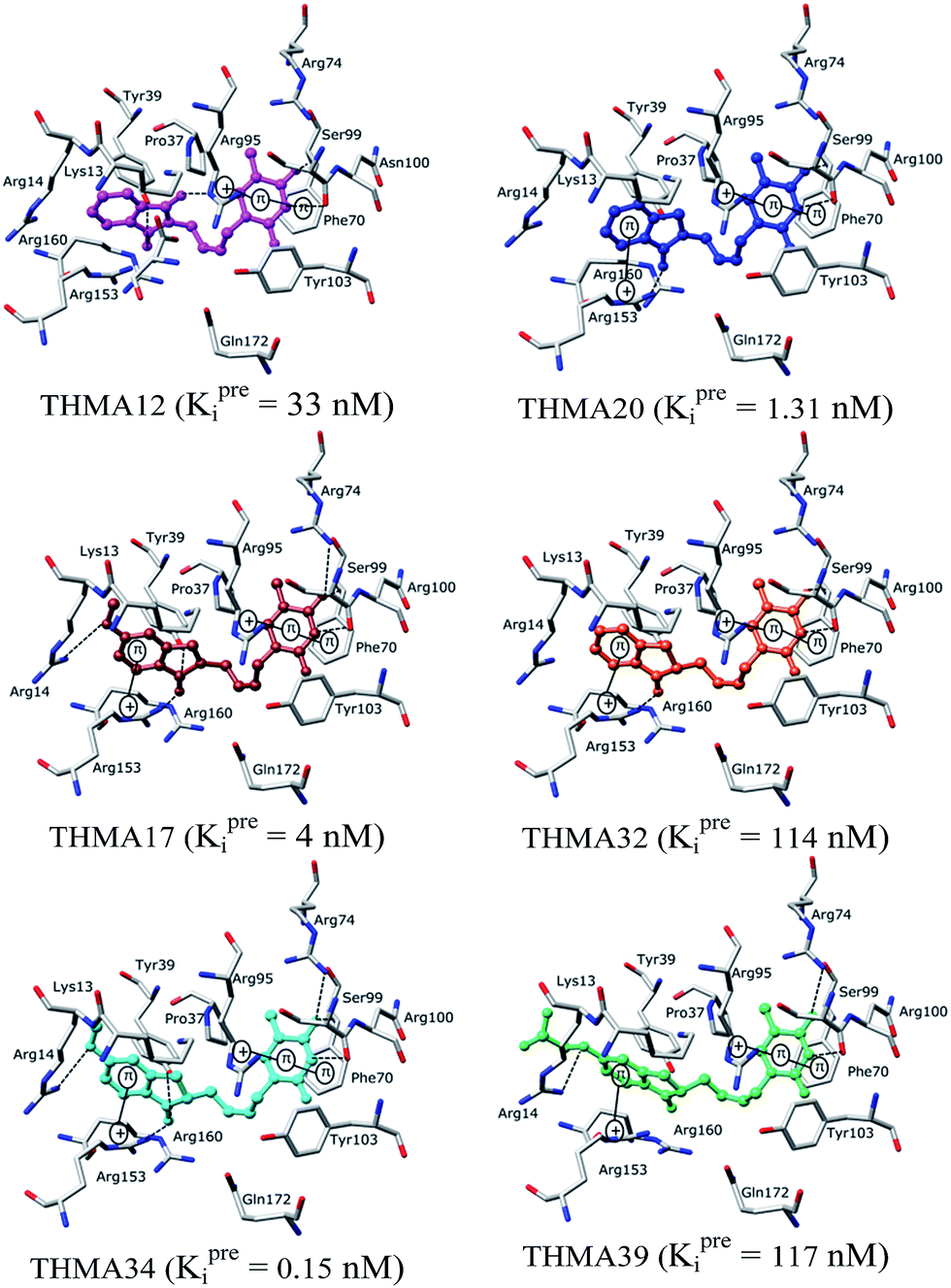
| | Fig. 10 Some of the most active designed analogs are shown at the active site of TMPKmt in stick representation. Hydrogen bonds are shown in black dashed lines, other important interactions are shown as solid black lines. | |
 |
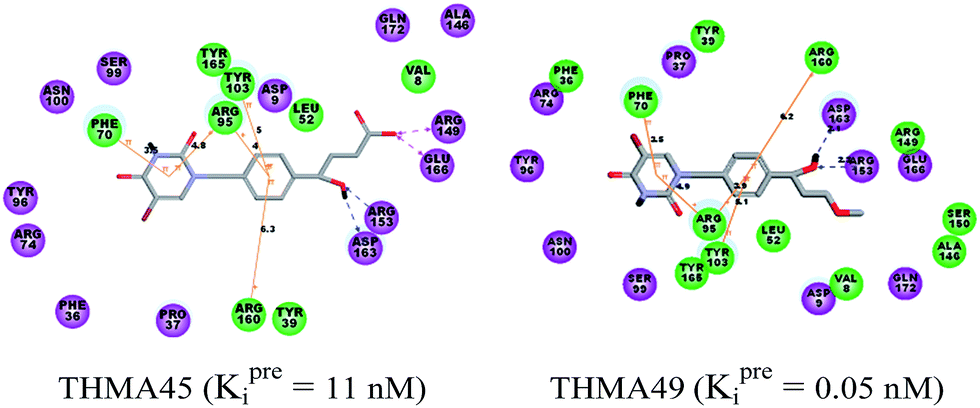
| | Fig. 11 2D depiction of interaction between TMPKmt and THMA45 with aromatic spacer at the enzyme's active site, cation–aromatic π and aromatic π–π interactions are in orange line, HBs are in dashed blue line and charge interactions in dashed purple line. | |
 |
| | Fig. 12 Interaction energy breakdown to residue contributions for the best designed analogs with aliphatic spacer (THMA34, red) and with aromatic spacer (THMA49, green). | |
The picomolar Kprei of some designed compounds may represent a somewhat too optimistic prediction of activity that falls outside of the concentration range of the training set inhibitors considered in the QSAR model of TMPKmt inhibition. However, the indication of elevated inhibitory potencies of these molecules can still be a useful hint for medicinal chemists developing more potent TMPKmt inhibitors to explore the proposed chemical structures.
New analogs with an aliphatic spacer.
Substitution in positions R4 to R7 of the naphtholactam moiety. These analogs THMA1–11 are derived from the most potent training set inhibitor THMD7 (Fig. 5) and are intended to involve Lys13 and Arg14 residues into inhibitor binding. The best of them THMA6, which bears two hydroxymethyl groups in the positions R5 and R6 reached predicted inhibitory potency towards the TMPKmt Kprei = 4 nM (Fig. 10).
Replacement of the naphtholactam by phthalimide moiety. Replacement of the bulky naphtholactam moiety by a smaller less hydrophobic phthalimide was not favourable for the analogue binding. Substitution with a methoxy group at position 4 in THMA17 resulted in a predicted Kprei of 4 nM. Surprisingly placement of a carboxyl group to the R4 position in THMA16 did not lead to the expected favourable interaction with Arg14. Despite the fact that the HB between the negatively charged carboxyl oxygen and Arg14 is conserved the potency remained almost unchanged (Kprei = 6 nM) with respect to THMA17, due in part to the COO(−) group's repulsive interaction with Glu124 and Glu166 in THMA16.
Removal of one ketone from phthalimido moiety. When the phthalimido moiety was replaced by an isoindoline the predicted activity increased almost 30 times c.f. THMA12 (Kprei = 33 nM) and THMA20 (Kprei = 1.31 nM), most probably due to unfavourable interaction of the ketone oxygen with the hydroxyl group of Tyr39 (Fig. 10). Additional substitutions at positions 3 and 5 by methyl and methylamide groups, respectively (THMA26) or alternatively by chlorines in positions R1 to R4 (THMA31) resulted in predicted subnanomolar inhibitory potencies of the designed analogs c.f. THMA27.
Removal of nitrogen atom from 5 membered isoindoline ring. When the isoindoline is replaced by an 1-indanone by replacement of the sp2 ring nitrogen with a sp3 methylene group the predicted activity of the analogs drops by two orders of magnitude, c.f. THMA20 (Kprei = 1.31 nM) and THMA32 (Kprei = 114 nM). However, this substitution confers higher flexibility and improved spatial orientation of the indanone moiety. Substitutions in position R5 by methoxy or acetamide groups led to analogs with predicted low nanomolar and subnanomolar inhibitory potencies (THMA34 and THMA43). The attachment of anionic carboxyl –COO(−) and especially methoxyacetate –OCH2COO(−) groups into the R5 position added two hydrogen bonds of the THMA39 to the residues Arg14 and Arg74 (Fig. 10), but resulted in a relatively weak predicted binding affinity due to unfavourable solvent effect and anionic inhibitor desolvation.
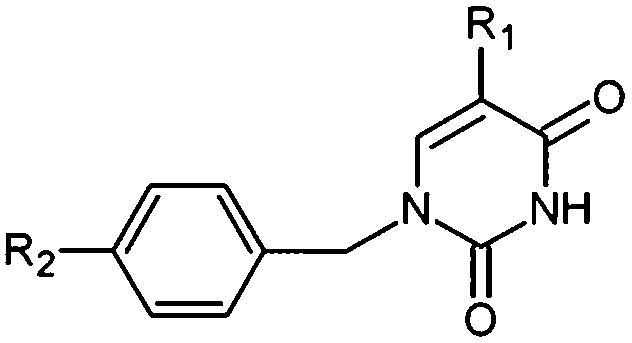
New analogs with aromatic spacer. The training set THMDs is composed with almost 50% aliphatic spacer (alTHMDs, 0.27 ≤ Kexpi ≤ 6 μM) and 50% aromatic spacer (arTHMDs, 10 ≤ Kexpi ≤ 202 μM). At first sight and according to their potency it seems obvious that designed arTHMAs will never reach the potency level of alTHMAs. The analysis of residues contribution to Eint breakdown for THMD13 (the best arTHMDs in the training set) in Fig. 7 shows that the three additional interactions between the aromatic spacer ring and residues Arg95 and Arg160 (cation–π) and Tyr103 (π–π) raise substantially the affinity towards TMPKmt. Moreover as depicted in 2D in Fig. 11 and the graph in Fig. 12 the carboxylic group repulsive interactions with Glu124 and Glu166 lowers the affinity (THMA45, 47 and 48). Oppositely the methoxy group enhances the THMAs stabilizing interactions with the enzyme exemplified by THMA49 and 50 (Fig. 13). Besides the basic stacking involving the thymine moiety common to both alTHMAs and arTHMAs, these three additional π–π and cation–π interactions and finally two HBs between the OH on R2 and Arg153 and Asp163 justify the significant increase of the predictive potency exhibited by arTHMAs.
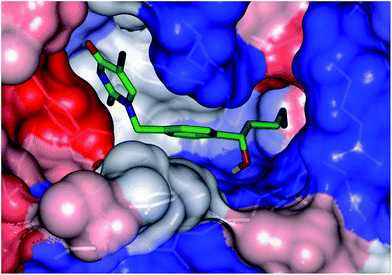 |
| | Fig. 13 Connolly surface of the predicted most active arTHMA THMA49 at the active site of TMPKmt. The binding cleft surface is coloured according to residue hydrophobicity: red-hydrophobic, blue-hydrophilic and white-intermediate residues. | |
The overall contribution to Eint of the residues specific for TMPKmt of 15% for THMA34 vs. 5% for THMA49 breakdown into residue contributions: Arg14 (7% – THMA34 vs. 0% – THMA49), Tyr39 (4% – THMA34 vs. 1% – THMA49) and Asn100 (4% – THMA34 vs. 4% – THMA49) render the best arTHMA less selective than its alTHMA counterpart. For arTHMAs the conversion of the carboxylic acid group (THMA45, Kprei = 8 nM) into a bulkier methyl ester resulted in a detrimental decrease of affinity (THMA46, Kprei = 458 nM).
3.5 ADME-related properties of THMA inhibitors
Drug likeness parameter (#stars, for definition see the Methods section) computed for known THMD inhibitors of TMPKmt, antituberculotics either in clinical use or in clinical trials (Table 6) and selected potent designed analogs THMAx (Table 7) characterizes the ADME related properties relevant for the pharmacokinetic profile of a compound. As we can see from Tables 6 and 7, the most potent THMDs as well as the predicted most active designed THMAs display favourable values of #stars parameter equal to 0 for all except one compound. Five of the novel antituberculosis drugs in trials also display #stars equal to zero. Therefore, the designed THMA analogs are predicted to possess favourable pharmacokinetic profiles and thus represent suitable candidates for synthesis and experimental testing.
Table 6 Predicted ADME-related properties of some THMDs and known antituberculotics either in use or in test computed by QikProp.30 (*) Star indicating that the property descriptor value falls outside the range of values for 95% of known drugs
| Compoundsa |
#starsb |
Mwc [g mol−1] |
Smold [Å2] |
Smol,hfoe [Å2] |
Vmolf [Å3] |
RotBg |
HBdonh |
HBacci |
logPo/wj |
logSwatk |
logKHSAl |
logB/Bm |
BIPCacon [nm s−1] |
#metao |
Kexpip [μM] |
HOAq |
%HOAr |
| Known compounds inhibitors with experimentally determined inhibition constants, see Table 1. Drug likeness, number of property descriptors (from 24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release 14) that fall outside the range of values for 95% of known drugs (range or recommended values: 0–5). Molecular weight in g mol−1 (range for 95% of drugs: 130–725 g mol−1).30 Total solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2). Hydrophobic portion of the solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2). Total volume of molecule enclosed by solvent-accessible molecular surface, in Å3 (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3). Number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds (range for 95% of drugs: 0–15). Estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can be non-integer (range for 95% of drugs: 0.0–6.0). Estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can be non-integer (range for 95% of drugs: 2.0–20.0). Logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6.5). Logarithm of predicted aqueous solubility, logS. S in mol dm−3 is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (range for 95% of drugs: −6.0 to 0.5). Logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5). Logarithm of predicted brain–blood partition coefficient. Note: QikProp predictions are for orally delivered drugs so, for example, dopamine and serotonin are CNS negative because they are too polar to cross the blood–brain barrier (range for 95% of drugs: −3.0 to 1.2). Predicted apparent Caco-2 cell membrane permeability in Boehringer-Ingelheim scale, in [nm s−1] (range for 95% of drugs: <25 poor, >500 great). Number of likely metabolic reactions (range for 95% of drugs: 1–8). Experimental THMD inhibitory activity.6,12,13 Human oral absorption (1-low, 2-medium, 3-high). Percentage of human oral absorption in gastrointestinal tract (<25% – poor, >80% high). |
| THMD6 |
1 |
392.4 |
622.1 |
176.5 |
1125.1 |
5 |
1 |
7.5 |
1.8 |
−4.1 |
0 |
−2.0 |
47.5 |
4 |
0.75 |
3 |
67 |
| THMD7 |
0 |
383.4 |
577.7 |
162.8 |
1071.7 |
4 |
1 |
8 |
1.7 |
−3.3 |
−0.2 |
−1.2 |
201.8 |
3 |
0.27 |
3 |
78 |
| THMD9 |
0 |
288.3 |
535.5 |
185.4 |
920.9 |
5 |
2 |
5.5 |
1.8 |
−3.3 |
−0.3 |
−1.7 |
20.3 |
4 |
55 |
2 |
61 |
| THMD11 |
0 |
301.3 |
573.1 |
215.8 |
988.8 |
6 |
3 |
6 |
0.7 |
−2.1 |
−0.5 |
−1.9 |
32.5 |
5 |
112 |
2 |
58 |
| THMD13 |
0 |
367.2 |
562.5 |
136.7 |
970.8 |
6 |
2 |
5.5 |
2.5 |
−3.9 |
−0.2 |
−1.6 |
24.2 |
3 |
10 |
2 |
66 |
| THMD15 |
0 |
288.3 |
535.1 |
133.9 |
921.4 |
6 |
2 |
5.5 |
1.8 |
−3.2 |
−0.3 |
−1.9 |
16.1 |
3 |
202 |
2 |
59 |
| Rifampin |
1 |
137.1 |
313.3 |
0.0 |
479.2* |
2 |
3 |
4.5 |
−0.7 |
0 |
−0.8 |
−0.8 |
267.5 |
2 |
— |
2 |
67 |
| Isoniazid |
4 |
123.1* |
299.3 |
0.0 |
442.6* |
1 |
2 |
5 |
−0.6 |
−0.5 |
−0.8 |
−0.7 |
298.4 |
4 |
— |
2 |
67 |
| Ethambutol |
2 |
204.3 |
475.7 |
395.8 |
805.7 |
11 |
4 |
6.4 |
−0.2 |
0.6 |
−0.8 |
0.0 |
107.8 |
4 |
— |
2 |
62 |
| Pyrazinamide |
10 |
823.0* |
1090* |
850.0* |
2300* |
25* |
6 |
20.35* |
3.0 |
−3.1 |
−0.3 |
−2.7 |
38.2 |
11* |
— |
1 |
34 |
| Gatifloxacin |
0 |
375.4 |
597.5 |
355.7 |
1093.0 |
2 |
1 |
6.8 |
0.5 |
−4.0 |
0 |
−0.6 |
17.0 |
1 |
— |
2 |
52 |
| Moxifloxacin |
0 |
401.4 |
641.2 |
395.6 |
1167.1 |
2 |
1 |
6.8 |
1.0 |
−4.7 |
0.2 |
−0.6 |
20.9 |
1 |
— |
2 |
56 |
| Rifapentine |
10 |
877.0* |
1024.3* |
844.9* |
2332.6* |
24* |
6 |
20.9* |
3.6 |
−2.2 |
−0.2 |
−1.5 |
224.0 |
13* |
— |
1 |
51 |
| Bedaquiline |
4 |
555.5 |
786.5 |
213.7 |
1531.7 |
9 |
1 |
3.8 |
7.6* |
−6.9 |
1.7 |
0.4 |
1562.2 |
5 |
— |
1 |
100 |
| Delamanid |
2 |
534.5 |
795.6 |
284.4 |
1469.9 |
7 |
0 |
6.0 |
5.8 |
−7.6 |
1.0 |
−1.0 |
590.9 |
2 |
— |
1 |
85 |
| Linezolid |
0 |
337.4 |
554.6 |
337.2 |
995.4 |
2 |
1 |
8.7 |
0.6 |
−2.0 |
−0.7 |
−0.5 |
507.0 |
2 |
— |
3 |
79 |
| Sutezolid |
1 |
353.4 |
594.0 |
330.6 |
1046.2 |
2 |
1 |
7.5 |
1.3 |
−3.4 |
−0.4 |
−0.4 |
449.3 |
0 |
— |
3 |
82 |
| Ofloxacin |
1 |
361.4 |
580.5 |
337.0 |
1044.0 |
1 |
0 |
7.3 |
−0.4 |
−2.8 |
−0.5 |
−0.4 |
25.9 |
1 |
— |
2 |
50 |
| Amikacin |
14 |
585.6 |
738.3 |
350.3 |
1499.5 |
22* |
17* |
26.9* |
−7.9* |
−0.2 |
−2.1 |
−3.5 |
0 |
14* |
— |
1 |
0 |
| Kanamycin |
10 |
484.5 |
655.8 |
258.9 |
1290.9 |
17* |
15* |
22.7* |
−6.7* |
2.0 |
−1.4 |
−3.1 |
0 |
12* |
— |
1 |
0 |
| Imipenem |
0 |
299.3 |
486.5 |
259.1 |
879.4 |
8 |
3 |
7.2 |
1.0 |
−1.8 |
−0.7 |
−1.4 |
35.0 |
3 |
— |
3 |
61 |
| Amoxicillin |
2 |
365.4 |
560.8 |
164.6 |
1032.9 |
6 |
4.25 |
8.0 |
−2.5 |
−0.8 |
−1.1 |
−1.5 |
1.0 |
5 |
— |
1 |
12 |
| Clavulanate |
0 |
199.2 |
396.1 |
184.6 |
629.5 |
4 |
2 |
6.5 |
−0.8 |
0.3 |
−1.3 |
−1.3 |
13.3 |
2 |
— |
2 |
42 |
Table 7 Predicted ADME-related properties of selected most potent designed THMDs analogues computed by QikProp30
| Novel analogsa |
#starsb |
Mwc [g mol−1] |
Smold [Å2] |
Smol,hfoe [Å2] |
Vmolf [Å3] |
RotBg |
HBdonh |
HBacci |
logPo/wj |
logSwatk |
logKHSAl |
logB/Bm |
BIPCacon [nm s−1] |
#metao |
Kpreip [nM] |
HOAq |
%HOAr |
| See Table 5 for the chemical structures of the virtual hits. Drug likeness, number of property descriptors (from 24 out of the full list of 49 descriptors of QikProp, ver. 3.7, release 14) that fall outside the range of values for 95% of known drugs (range or recommended values: 0–5). Molecular weight in g mol−1 (range for 95% of drugs: 130–725 g mol−1).30 Total solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2). Hydrophobic portion of the solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2). Total volume of molecule enclosed by solvent-accessible molecular surface, in Å3 (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3). Number of non-trivial (not CX3), non-hindered (not alkene, amide, small ring) rotatable bonds (range for 95% of drugs: 0–15). Estimated number of hydrogen bonds that would be donated by the solute to water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can be non-integer (range for 95% of drugs: 0.0–6.0). Estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution. Values are averages taken over a number of configurations, so they can be non-integer (range for 95% of drugs: 2.0–20.0). Logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6.5). Logarithm of predicted aqueous solubility, logS. S in mol dm−3 is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (range for 95% of drugs: −6.0 to 0.5). Logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5). Logarithm of predicted brain–blood partition coefficient. Note: QikProp predictions are for orally delivered drugs so, for example, dopamine and serotonin are CNS negative because they are too polar to cross the blood–brain barrier (range for 95% of drugs: −3.0 to 1.2). Predicted apparent Caco-2 cell membrane permeability in Boehringer-Ingelheim scale, in [nm s−1] (range for 95% of drugs: <25 poor, >500 great). Number of likely metabolic reactions (range for 95% of drugs: 1–8). Predicted Kpreis were estimated from the QSAR equation, Table 3. Human oral absorption (1-low, 2-medium, 3-high). Percentage of human oral absorption in gastrointestinal tract (<25% – poor, >80% high). |
| THMA6 |
0 |
407.4 |
634.3 |
245.4 |
1174.3 |
8 |
3 |
9.9 |
1.1 |
−3.2 |
−0.3 |
−2.0 |
70.6 |
5 |
3 |
3 |
66 |
| THMA12 |
0 |
325.3 |
546.4 |
170.1 |
971.3 |
4 |
1 |
6.5 |
1.7 |
−3.4 |
−0.1 |
−1.2 |
207.2 |
3 |
21 |
3 |
79 |
| THMA28 |
0 |
341.4 |
590.1 |
290.9 |
1058.0 |
5 |
1 |
7.3 |
2.0 |
−3.6 |
−0.1 |
−1.2 |
341.5 |
5 |
33 |
3 |
84 |
| THMA32 |
0 |
310.4 |
571.3 |
216.4 |
1002.9 |
4 |
1 |
5.5 |
2.5 |
−4.1 |
0.1 |
−1.1 |
379.2 |
5 |
77 |
3 |
88 |
| THMA34 |
0 |
384.4 |
639.2 |
309.7 |
1147.6 |
6 |
2 |
8.3 |
2.0 |
−4.1 |
−0.3 |
−2.0 |
17.2 |
6 |
0.094 |
2 |
61 |
| THMA39 |
0 |
384.4 |
669.8 |
262.1 |
1174.7 |
7 |
2 |
8.3 |
1.9 |
−4.4 |
−0.3 |
−2.6 |
7.1 |
7 |
101 |
2 |
54 |
| THMA49 |
0 |
369.2 |
557.8 |
200.4 |
976.7 |
7 |
2 |
6.9 |
1.9 |
−3.3 |
−0.3 |
−1.0 |
396.6 |
3 |
0.03 |
3 |
85 |
| THMA50 |
0 |
304.3 |
560.9 |
286.1 |
983.8 |
7 |
2 |
6.9 |
1.7 |
−3.0 |
−0.3 |
−1.2 |
426.5 |
4 |
0.04 |
3 |
84 |
The most active designed alTHMAs (THMA6 and 34) and arTHMAs (THMA49 and 50) display pharmacokinetic profiles for almost all descriptors considered (Table 7).30 There is, however, a noticeable difference in the cell wall permeability parameter BIPCaco-2 (Table 7) showing that arTHMAs display higher predicted permeability rates than the two alTHMAs. It is worthwhile mentioning that about 50% of new antituberculotics in (pre)clinical trials fall into low BIPCaco-2 permeability range. arTHMAs' predicted percentage of human oral absorption in gastrointestinal tract (HOA) falls into the range of 85% against 66% for alTHMAs, which suggest the possibility of higher oral bioavailability for the most active arTHMAs.30 According to these descriptors the arTHMAs analogs display more favourable predicted ADME-related profiles and drug-like properties and thus represent candidate molecules suitable for synthesis.
4. Conclusions
The high statistical significance of the QSAR model and PH4 pharmacophore model derived from a training set of 20 and validation set of 3 THMD compounds was well documented. Presence of the naphtholactam moiety in the most potent alTHMAs TMPKmt inhibitors and analysis of the active site residue contributions to Eint directed us in our effort to optimize the structures of the known inhibitors and enhance the involvement of the residues Lys13 and Arg14 in the inhibitor binding. We have replaced in alTHMAs the bulky and rigid naphtholactam moiety by smaller less hydrophobic and more flexible phthalimide, isoindoline and indanone moieties. Additional substitutions with methoxy, hydroxy-methyl and acetamide groups in positions R4 and R5 of the aromatic scaffolds improved the binding site interactions by specific hydrogen bonding involving the Lys13, Arg14, Arg153 and Tyr39 residues to reach 0.15 nM predicted potency. These residues were identified and targeted based on the active site interaction energy Eint breakdown for a molecular mechanics all-atom model of inhibitor–TMPKmt binding and comparative analysis, which led to identification of new potent inhibitor candidates. In the same way replacement of the butenyl spacer of alTHMAs by an aromatic one in arTHMAs intensifies the affinity by adding three π–π and cation–π interactions and two HBs to reach picomolar predicted potency.
Although our activity predictions may be somewhat too optimistic, they still point to a specific subset of the chemical space which may contain subnanomolar TMPKmt inhibitors with favourable pharmacokinetic profiles. A more systematic search through screening of combinatorial library targeted around our designed inhibitors THMA34, THMA27, THMA48 and THMA49 and subsequent activity evaluation in an enzymatic assay may lead to a discovery of new potent antituberculotics.
Notes and references
- C. Dye, D. Maher and D. Weil, et al., Int. J. Tuberc. Lung Dis., 2006, 10, 460–462 CAS.
- A. Zumla, M. Raviglione, R. Hafner and F. von Reyn, N. Engl. J. Med., 2013, 368, 745–755 CrossRef CAS PubMed.
- World Health Organization, Global tuberculosis report, Geneva, 2012, http://www.who.int/tb/publications/global_report/en/ Search PubMed.
- A. Haouz, V. Vanheusden and H. Munier-Lehmann, et al., J. Biol. Chem., 2003, 278, 4963–4971 CrossRef CAS PubMed.
- V. Vanheusden, P. V. Rompaey and H. Munier-Lehmann, et al., Bioorg. Med. Chem. Lett., 2003, 13, 3045–3048 CrossRef CAS PubMed.
- O. Familiar, H. Munier-Lehmann and A. Negri, et al., ChemMedChem, 2008, 3, 1083–1093 CrossRef CAS PubMed.
- B. Gopalakrishnan, V. Aparna and J. Jeevan, et al., J. Chem. Inf. Model., 2005, 45, 1101–1108 CrossRef CAS PubMed.
- T. Ursby, M. Weik and E. Fioravanti, et al., Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 607–614 CrossRef.
- I. Li de la Sierra, H. Munier-Lehmann and A. M. Gilles, et al., J. Mol. Biol., 2001, 311, 87–100 CrossRef CAS PubMed.
- V. Frecer, P. Seneci and S. Miertus, J. Comput.-Aided Mol. Des., 2011, 25, 31–49 CrossRef CAS PubMed.
- L. C. Owono Owono, M. Keita and E. Megnassan, et al., Tuberc. Res. Treat., 2013, 1–13 CrossRef PubMed.
- S. Pochet, L. Dugue and G. Labesse, et al., ChemBioChem, 2003, 4, 742–747 CrossRef CAS PubMed.
- C. Gasse, D. Douguet and V. Huteau, et al., Bioorg. Med. Chem., 2008, 16, 6075–6085 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook and G. Z. Feng, et al., Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Discovery Studio molecular modeling and simulation program, version 2.5, Accelrys, Inc., San Diego, CA, 2009 Search PubMed.
- V. Frecer, S. Miertus, A. Tossi and D. Romeo, Drug Des. Discovery, 1998, 15, 211–231 CAS.
- V. Frecer and S. Miertus, Macromol. Chem. Phys., 2002, 203, 1650–1657 CrossRef CAS.
- V. Frecer, F. Berti, F. Benedetti and S. Miertus, J. Mol. Graphics Modell., 2008, 27, 376–387 CrossRef CAS PubMed.
- B. Dali, M. Keita and E. Megnassan, et al., Chem. Biol. Drug Des., 2012, 79, 411–430 CAS.
- E. Megnassan, M. Keita and C. Bieri, et al., Med. Chem., 2012, 8(5), 970–984 CrossRef CAS.
- M. K. Gilson and B. Honig, J. Comput.-Aided Mol. Des., 1991, 5, 5–20 CrossRef CAS PubMed.
- W. Rocchia, S. Sridharan and A. Nicholls, et al., J. Comput. Chem., 2002, 23, 128–137 CrossRef CAS PubMed.
- C. J. F. Böttcher, Theory of Electric Polarization, Elsevier Press, Amsterdam, 1973 Search PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- V. Frecer and S. Miertus, Int. J. Quantum Chem., 1992, 42, 1449–1468 CrossRef CAS.
- S. Fischer, J. C. Smith and C. Verma, J. Phys. Chem. B, 2001, 105, 8050–8055 CrossRef CAS.
- S. M. Schwarzl, T. B. Tschopp, J. C. Smith and S. Fischer, J. Comput. Chem., 2002, 23, 1143–1149 CrossRef CAS PubMed.
- J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2006, 49, 5880–5884 CrossRef CAS PubMed.
- H. Li, J. Sutter and R. Hoffmann, in Pharmacophore Perception, Development and Use in Drug Design, ed. O. F. Güner, International University Line, La Jolla, CA, USA, 2000, pp. 171–189 Search PubMed.
- QikProp, version 3.7, release 14, X Schrödinger, LLC, New York, NY, 2014 Search PubMed.
- E. M. Duffy and W. L. Jorgensen, J. Am. Chem. Soc., 2000, 122, 2878–2888 CrossRef CAS.
- W. L. Jorgensen and E. M. Duffy, Bioorg. Med. Chem. Lett., 2000, 10, 1155–1158 CrossRef CAS PubMed.
- W. L. Jorgensen and E. M. Duffy, Adv. Drug Delivery Rev., 2002, 54, 355–366 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O
O
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2CH2OH
CCH2CH2OH