
A strategy to design biocompatible polymer particles possessing increased loading efficiency and controlled-release properties
Phakkhananan Pakawanitab,
Supon Anantab,
Tae Kwan Yunc,
Jae Young Bae*c,
Wongi Jangd,
Hongsik Byun*d and
Jun-Hyun Kim*a
aDepartment of Chemistry, Illinois State University, Normal, Illinois 61790-4160, USA. E-mail: jkim5@ilstu.edu
bDepartment of Physics and Materials Science, Chiang Mai University, Chiang Mai, 50200, Thailand
cDepartment of Chemistry, Keimyung University, Daegu, 704-701, South Korea. E-mail: jybae@kmu.ac.kr
dDepartment of Chemical System Engineering, Keimyung University, Daegu, 704-701, South Korea. E-mail: hsbyun@kmu.ac.kr
First published on 18th August 2014
Abstract
Temperature-responsive poly(N-isopropylacrylamide), or poly(NIPAM), layers were reliably prepared around guest molecule (i.e., rhodamine B)-loaded mesoporous silica (SiO2) particles via thermally- and light-induced radical polymerizations. Subsequent removal of the sacrificial SiO2 particles with dilute hydrofluoric acid led to the formation of biocompatible polymer particles possessing a high dose of rhodamine B. The use of SiO2 core templates not only led to the formation of a uniform coating of the poly(NIPAM) layers, but also increased the stability of the guest molecule, rhodamine B, throughout polymerization. Interestingly, the light-induced radical polymerization method resulted in much less inevitable leaching and decomposition of azo-based guest molecules. The structural information and overall dye-loading efficiency of the mesoporous particles and the final polymer particles were then thoroughly examined by electron microscopes, dynamic light scattering, and fluorescence spectroscopy. As poly(NIPAM)-based particles exhibited significant swelling and deswelling properties above and below the lower critical solution temperature, the controlled-release properties of the poly(NIPAM) particles prepared by both methods were also evaluated. Generally, the dye-loaded poly(NIPAM) particles prepared by the light-induced approach resulted in a thinner coating of the polymer layers and exhibited much higher loading and tunable release profiles of the loaded guest molecules than those prepared by the thermally-induced polymerization. Given these features, the generalization of our strategy to design chemotherapeutically interesting drug-loaded polymer particles that are biocompatible and sensitive to external stimuli will allow for the further development of novel biomedical delivery and treatment systems.
Introduction
Over the last few decades, stimuli-responsive polymer-based delivery materials possessing a moderate guest molecule-loading ability and controlled-release properties have been designed to provide enhanced biodistribution, pharmacokinetics, and efficacy for bio-imaging/sensing and therapeutic applications.1–9 Biocompatible poly(N-isopropylacrylamide), or poly(NIPAM), derivatives are especially interesting materials due to their tunable structural volume changes at the physiological temperature and pH as well as their easy preparation by conventional radical polymerization.3,5–7,10–18 These polymer particles as delivery vehicles can further be tailored to reliably form hollow-type poly(NIPAM) particles utilizing sacrificial silica (SiO2) particles.4,15,16 The resulting polymer particles can then exhibit a much higher loading capability of guest molecules and their rapid controlled-release properties caused by the significant volume changes to external stimuli (e.g., temperature, pH, and light) make them ideal for delivery applications.Mesoporous SiO2 particles as a sacrificial core template have been widely explored as drug cargo due to their large surface area, tunable pore sizes, homogeneous size distribution, and easy surface modification.15,19–23 These particles not only allow for a wide range of drug payloads, but also serve as a great core template to be coated with organic, inorganic, and polymeric materials. Particularly, the loading of guest molecules within mesoporous SiO2 particles can be simply achieved by mixing them in the appropriate solvents overnight. The surface of these dye-loaded SiO2 particles can be readily modified with organic molecules to prevent the leaching of the loaded guest molecules during the extensive purification and polymerization steps. Furthermore, after being coated with polymeric shell materials, these mesoporous particles can be dissolved with a dilute hydrofluoric acid solution,4,20 while a high dose of the loaded molecules remain within the shell layers. The removal of the SiO2 core particles leaves only the biocompatible polymer and the guest molecules behind, which can minimize the potential toxicity of these materials as drug-delivery vehicles.4,24 In this study, mesoporous SiO2 particles were initially prepared with a high dose of rhodamine-B cargo, and the surfaces of these particles were then modified with MPS (3-trimethoxysilyl propyl methacrylate) to minimize the leakage of the loaded dye. As the MPS surface modifier also possesses unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moieties,2,16,20,25 the dye-loaded SiO2 particles were subsequently subjected to the radical polymerization of NIPAM, followed by the removal of the SiO2 particles, leaving the highly-loaded dye molecules within the polymer structures.
C moieties,2,16,20,25 the dye-loaded SiO2 particles were subsequently subjected to the radical polymerization of NIPAM, followed by the removal of the SiO2 particles, leaving the highly-loaded dye molecules within the polymer structures.
The formation of a poly(NIPAM) shell around the aforementioned dye-loaded mesoporous SiO2 particles can be accomplished by thermally- and light-induced radical polymerizations. As the thermally-induced polymerization approach often employs organic peroxide radical initiators (e.g., ammonium persulfate, APS) at a relatively mild reaction temperature (e.g., 70 °C),4,16,26 our dye-loaded SiO2 particles may induce the leaching of the dye molecules during a long period of polymerization. In addition, it is reported that azo dyes (including rhodamine B and methylene blue) can undergo decomposition and decolorization in the presence of persulfate-based radical sources (enhanced decomposition with an increased reaction temperature),27,28 which can further lower the guest molecule-loading efficiency within the delivery vehicles. As such, we attempt to grow poly(NIPAM) layers around the dye-loaded SiO2 core particles via solar-simulated light-induced radical polymerization at room temperature to decrease the thermally-induced leakage and to avoid the notable decomposition of the guest molecules during polymerization. This light-induced radical polymerization can be achieved in the presence of readily available organic acids (e.g., oxalic acid) as photo-initiators, which can effectively induce the polymerization of vinyl monomers at room temperature under light irradiation.29–33 The previous study based on the light-induced polymerization of methyl methacrylate with the initiator azobisisobutyronitrile at room temperature showed the formation of a more uniform polymer with a lower molecular weight than that formed from the thermally-induced polymerization at a high reaction temperature;33 this may offer the additional advantage of precisely controlling the coating of poly(NIPAM) layers around the SiO2 core particles. To the best of our knowledge, this is the first time demonstration that light-induced polymerization using commonly-available oxalic acid as the photo-initiator reliably forms temperature-responsive polymer layers around guest molecule-loaded core particles. The SiO2 core particles are then carved out with a dilute HF solution to form hollow-type poly(NIPAM) particles containing rhodamine B as a model drug. It is known that biocompatible polymer particles consisting of poly(NIPAM)-based materials can readily undergo significant thermally-induced structural volume changes at the lower critical solution temperature (LCST), leading to a tunable triggered-release property.2–4,10,34 Investigating unique transition of poly(NIPAM) particles possessing the ability of loading and release a high density of guest molecules (e.g., drugs or combinations of drugs) can allow for the development of interesting temperature-responsive materials for various biological applications. Our approach can be easily generalized as it utilizes the readily available poly(NIPAM) around the sacrificial mesoporous SiO2 particles as a template of hollow-core structures with a high loading capability and external stimuli-responsive triggered-release properties. The surface of these polymer-based carriers can be further modified with biologically specific/selective entities for the development of targeted drug-delivery systems.
Experimental section
Materials
The N-isopropylacrylamide (NIPAM, 99%) monomer was recrystallized in hexane and completely dried under vacuum prior to use. The tetraethyl orthosilicate (TEOS, 98%), cetyltrimethylammonium chloride (CTACl, Aldrich, 25 wt.%), aqueous ammonia (25–30%), ammonium persulfate (APS, 98%), N,N′-methylenebisacrylamide (BIS, 96%), 3-(trimethoxysilyl)propyl methacrylate (MPS, 98%), ethyl alcohol (HPLC grade), rhodamine B dye (98%), oxalic acid anhydrous (99%) and hydrofluoric acid (HF, 49%) were used as received without any further purification. The water used in all reactions was purified to a resistance of 18 MΩ (Nanopure water system; Barnstead/Thermolyne) and filtered through a 0.2 μm membrane to remove any impurities. All glassware was cleaned with a base bath and then rinsed with pure water.Preparation of mesoporous silica (SiO2) particles
Mesoporous SiO2 particles were synthesized using a cationic surfactant as the molecular template in an alkaline solution.35 The typical procedure involved the dissolution of CTACl (4.21 g) in DI water (250 mL) containing ethanol (150 mL) and aqueous ammonia (2.5 mL) by vigorous stirring. Once the color of the solution became clear, TEOS (2.5 mL) was added dropwise under constant stirring. After stirring the solution for an additional 24 hours at room temperature, the resulting SiO2 particles in the milky white solution were filtered to remove any unreacted starting materials, washed several times with DI water, and dried under vacuum. The dried SiO2 particles were calcined at 600 °C for 5 hours to remove the CTACl organic template, resulting in the formation of mesoporous SiO2 particles.Loading of rhodamine B within the mesoporous SiO2 particles and subsequent surface-modification of the dye-loaded mesoporous SiO2 particles with MPS
A series of glass vials containing the dried mesoporous SiO2 particles (0.10 g) were added to 10 mL of ethanol, and the SiO2 particles were completely suspended in the solution by sonication and stirring. Various concentrations of rhodamine B (5–100 wt%) were added to the vials under vigorous stirring. These mixture solutions were stirred for additional 6 hours to maximize the loading of the dye within the mesoporous SiO2 particles. Subsequently, MPS (18 μL, 0.07 mmol) was quickly introduced into these solutions using a gas-tight syringe and the resulting mixtures were stirred at room temperature overnight to ensure the surface modification. The coating of MPS around the surface of the SiO2 particles can lower the leaching of the loaded rhodamine B and serve as the linker when the polymer layers are coated around the SiO2 particles. An aliquot of the final mixture solution (1 mL) was diluted in 49 mL water and purified by centrifugation (3000 rpm for 15 minutes × 5, Sorvall Legend X1 Centrifuge Series) until the top solution became clear, indicating the successful removal of the free rhodamine B and any other impurities. The precipitated samples (i.e., the dye-loaded SiO2 particles) exhibited a bright red/pink color and were re-suspended in pure water (1 mL) prior to use.Poly(NIPAM) coating around dye-loaded mesoporous SiO2 particles via thermally- and light-induced polymerizations
Temperature-responsive poly(NIPAM) layers were prepared around the rhodamine-B-loaded mesoporous SiO2 particles via both thermally- and optically-induced radical polymerizations. Specifically, a purified solution of MPS-modified mesoporous SiO2 particles containing rhodamine B (1.0 mL) was diluted in pure water (98 mL) in a two-necked round bottom flask equipped with a reflux condenser and an inlet for argon gas. The monomer NIPAM (100 mg, 0.88 mmol), cross-linker BIS (10.0 mg, 0.065 mmol), and initiator APS (1.0 mg in 1 mL water) were added to the solution. This mixture solution was briefly sonicated and was purged with argon for 1 hour to remove any oxygen. The reaction flask was then heated to 70 °C in an oil bath and was vigorously stirred for the duration of the reaction (∼6 hours) to complete the polymerization. Similarly, solar-simulated light-induced radical polymerization was achieved using oxalic acid as the photo-initiator (0.5 mg in 1 mL water). As soon as oxalic acid was added to the quartz reaction flask containing the dye-loaded SiO2 particles, NIPAM, and BIS, the flask was completely wrapped in aluminum foil to prevent the random polymerization caused by any light source. After purging the reaction mixture with argon for 1 hour, the aluminum foil was removed from the reaction flask. Subsequently, the quartz reaction flask was exposed to a solar-simulated light for ∼6 hours at room temperature. The intensity of the light used was closely adjusted to the one-sun condition (∼100 mW cm−2, measured by the hand-held optical power meter-1916C, Newport Corp.), and there was a slight light-induced heating of the reaction flask throughout the polymerization period. Argon gas was bubbled through the solution for the duration of the polymerization. After the completion of the reactions, an aliquot of the final solution (5 mL) was centrifuged at 2000 rpm for 25 minutes twice. The supernatant was separated to remove any unreacted materials, free polymer nanoparticles, and leached rhodamine B. The precipitated samples were then re-suspended in pure water (5 mL) and stored at room temperature for later analysis. In our separate study, the light-induced polymerization using a higher concentration of the NIPAM monomer, the BIS cross-linker, and the oxalic acid initiator was performed in the absence of the dye-loaded SiO2 particles; the color of the resulting final solution was changed from colorless to milky white upon heating the sample well above the LCST of poly(NIPAM) (≥40 °C), clearly indicating the formation of a polymer.Loading and release of rhodamine B within mesoporous SiO2 and poly(NIPAM) particles prepared by thermally- and light-induced polymerizations
To examine the amount of loaded rhodamine B within the MPS-modified mesoporous SiO2 particles, an aliquot of the purified SiO2 solution (20 μL) was diluted in water (2 mL) and then concentrated HF (20 μL) was added to completely dissolve the SiO2 particles. After a brief sonication, the resulting solution was then subjected to the fluorescence spectroscopy measurement. The fluorescence intensities were compared to those of known concentrations of rhodamine B in an aqueous solution (i.e., the standard curve of rhodamine B in water) via the Beer–Lambert law to estimate the entirely encapsulated dye within the SiO2 particles. Although the fluorescence intensities of pure rhodamine B in an aqueous solution slightly decreased upon the addition of HF, the intensities of the dye-loaded mesoporous SiO2 particles slightly increased with the HF addition. These discrepancies were carefully normalized for all fluorescence measurements.Similarly, the loaded amount of rhodamine B within the poly(NIPAM) particles after polymerization, an aliquot of solution (10 mL) after both radical polymerizations, was centrifuged at 5000 rpm for 30 minutes to separate the leaked/free rhodamine B (top solution) and poly(NIPAM)-coated mesoporous SiO2 particles containing rhodamine B (bottom precipitates). The fluorescence of the top solution was subtracted from the total amount of rhodamine B loaded within the SiO2 particles, presumably representing the remaining dye within the poly(NIPAM)-coated mesoporous SiO2 particles. The bottom precipitates were then re-suspended in pure water (10 mL) and were placed in an ice bath prior to treatment with HF (100 μL) to completely dissolve the SiO2 particles, resulting in the complete loading of the dye within the poly(NIPAM) particles (considered as the total amount of rhodamine B within the polymer particles). Upon the removal of the SiO2 particles, the fluorescence intensity of the poly(NIPAM) particles notably increased, clearly implying these polymer particles were optically transparent for the encapsulated dye.36 These fluorescence results were still lower than the expected total amount of the loaded dye within the poly(NIPAM) particles, probably due to the decolorization/decomposition of rhodamine B by the radical initiators during polymerization. Although the loss of the fluorescence intensities could also be attributed to the partial quenching of locally concentrated rhodamine B within the polymer, we speculated that the major loss of the fluorescence was caused by the decolorization/decomposition of rhodamine B under such a low/diluted concentration of rhodamine B. These samples were then transferred to a dialysis bag (12–14 kDa MWCO, Spectra/Por 4 RC) and were subjected to the examination of the controlled-release properties upon heating (above the LCST) in a water bath by the fluorescence measurements. Similarly, the poly(NIPAM)-coated mesoporous SiO2 particles containing rhodamine B (i.e., without the removal of the SiO2 particles) were also tested for the fluorescence measurements upon heating in a water bath.
Characterization method
We used small-angle X-ray diffraction (XRD) and adsorption–desorption isotherms to determine the phase formation and specific surface area of the powdered mesoporous SiO2 particles.The powder of the mesoporous SiO2 particles was examined by XRD (an Empyrean instrument, PANalytical, MA) operating at a low angle (2θ from 0.3° to 10°) with a Cu target at 40 kV and 25 mA, using a speed of 2° min−1 and a step of 0.013°. The specific surface area (SBET) of the mesoporous SiO2 particles was determined by the adsorption–desorption isotherms of N2 at −196 °C (QUADRASORB SI adsorption apparatus, Quantachrome instrument, Boynton Beach, Florida). Prior to analysis, the sample was degassed at 200 °C for 2 hours under vacuum. The surface area of the sample was calculated by the linear part of the Brunauer–Emmett–Teller (BET) method based on the N2 adsorption isotherm data. The total pore volume (Vt) was then calculated from the amount adsorbed at a relative pressure (P/P0) of about 0.99. The pore size distribution was evaluated using the Barrett–Joyner–Halenda (BJH) method.
We utilized a combination of scanning electron microscopy (SEM), transmission electron microscopy (TEM), and dynamic light scattering (DLS) to characterize the size, morphology, and hydrodynamic diameters of the mesoporous SiO2 particles, MPS-modified mesoporous SiO2 particles containing rhodamine B, and poly(NIPAM)-coated mesoporous SiO2 particles before and after the removal of the SiO2 particles.
Particle diameter, uniformity, and overall morphology studies were performed using SEM (FEI-Quanta 450 operating at 20 kV) and TEM (Hitachi 8100 with an accelerating voltage of 200 kV and Hitachi H-7100 with an accelerating voltage of 100 kV). For the SEM images, all samples were deposited on silicon wafers, and completely dried at room temperature overnight before analysis. The samples were then coated with a thin Au film using a metal sputter coater (DESKII) under vacuum to avoid charging of the particles. For the TEM images, all samples were deposited on 300 mesh carbon-coated copper grids, and then dried overnight before analysis. These same samples were subjected to energy dispersive X-ray (EDX) analysis for the poly(NIPAM)-coated mesoporous SiO2 particles before and after the removal of the SiO2 core particles.
DLS (ZetaPALS, Brookhaven Instruments Corp.) equipped with a 35 mW solid state laser was used to measure the hydrodynamic diameters. All samples were suspended in pure water. The data reported represent an average of three measurements (100 s). The swelling and deswelling behavior of all samples was monitored when the temperature of the solution was adjusted below 25 °C and above 40 °C. In order to remove the SiO2 cores, the poly(NIPAM)-coated mesoporous SiO2 particles containing rhodamine B (30 μL in 2 mL water) was treated with a 20% HF solution (20 μL) and briefly sonicated for 5 minutes prior to analysis.
To determine the efficient loading/release of rhodamine B within the mesoporous SiO2 and poly(NIPAM) polymer particles, the florescence (luminescence spectroscopy LS55, Perkin Elmer) of various concentrations of rhodamine B in an aqueous solution was recorded and used as the calibration curve (ranging from 0.04 to 1.42 μg mL−1). As the fluorescence of rhodamine B was self-quenched at a high concentration, all samples were largely diluted and transferred to a four-sided plastic cuvette. The excitation and emission wavelengths were fixed in the setup at 550 nm and 581 nm. We noted that neither oxygen nor pH affected the rhodamine B spectra under our measurement conditions.
Results and discussion
Based on our previous study,16 we found a simple method to form stimuli-responsive polymer layers around sacrificial SiO2 cores via thermally-induced radical polymerization, followed by the removal of the SiO2 cores to prepare hollow polymer nanoparticles. The resulting particles exhibited significant structural volume changes as a function of the temperature and pH. Here, we took it one step further to demonstrate the efficient loading of rhodamine B dye as a model drug within mesoporous SiO2 particles and the subsequent coating of temperature-responsive polymer layers; this was followed by the removal of the SiO2 particles, leaving the loaded rhodamine B dyes within the polymer particles. These were then examined for the triggered-release property as a function of time.Initially, uniform mesoporous SiO2 particles were reliably prepared by a slight modification of the conventional method using a cationic surfactant in an alkaline solution (Fig. 1).35 TEM images and DLS analyses showed that the mesoporous particles were ∼450 nm and ∼480 nm in diameter with a very low polydispersity index (PDI: < 0.05), respectively. We observed arrays of hexagonal pores that formed a radial pattern from the center toward the outside of the spheres. It was assumed that the parallel alignment of the surfactants at the surface of the particles leads to a radially-aligned mesoporous structure. This observation was also consistent with the results of the XRD and N2 adsorption–desorption measurements described below. From the small-angle XRD pattern of the mesoporous SiO2 particles, the intense diffraction near 2θ's value of 2.59° suggested a (100) plane with highly ordered hexagonal mesoporous SiO2 materials. The major Bragg diffraction from the particles occurred at the slightly higher angle than that reported for characteristic hexagonal mesoporous SiO2 particles,37 which may indicate shorter distances between the pore centers than those of typical mesoporous SiO2 materials.
 | ||
| Fig. 1 TEM images and small-angle XRD pattern of mesoporous SiO2 particles. | ||
Fig. 2 shows the N2 adsorption–desorption isotherm of the mesoporous SiO2 particles. The particles exhibited features close to those of the IUPAC type I,38 but had a slight shoulder peak below the relative pressure of 0.3, indicating that the pore size is in an intermediate range between the micropore and mesopore regions. The isotherm of the particles displayed a well-defined capillary condensation step at a low relative pressure, P/P0 = 0.3. Unlike typical mesoporous SiO2 particles, the positions of the step-like curves were slightly shifted to lower values, presumably due to the presence of smaller pores within our SiO2 particles. The average pore size was then calculated from the desorption data using the BJH mode, and the specific surface area was obtained using the BET method in the P/P0 range of 0.05–0.30 (the overall textural characteristics of our mesoporous SiO2 particles are summarized in Fig. 2).
 | ||
| Fig. 2 The N2 adsorption–desorption isotherm of mesoporous SiO2 particles (SBET: BET surface area, Vt: total pore volume, DBJH: average pore diameter). | ||
Fig. 3 shows a rhodamine B dye loading process within the mesoporous SiO2 particles and the subsequent surface modification of the dye-loaded particles with MPS (3-(trimethoxysilyl)propyl methacrylate). The capping MPS molecules around the SiO2 surface via self-assembly could reduce the leaching of the rhodamine B dye from the SiO2 particles and the methacrylate moiety of MPS can serve as a linker to the polymer layers when radical polymerization initiates. After multiple centrifugation steps to remove the free dye molecules and unreacted MPS, the precipitates with a bright pink color (digital photo in Fig. 3) clearly showed the successful loading of rhodamine B within the SiO2 particles. Various dye-loaded SiO2 particles were then diluted and treated with HF (the dotted arrow in Fig. 3) to dissolve the SiO2 particles, which allowed for the estimation of the total amount of the loaded dye within the SiO2 particles by fluorescence measurements. The presentative fluorescence spectra of the rhodamine B-loaded SiO2 particles before and after the HF treatment are also shown in Fig. 3. Notable increase of both absorption and emission intensities clearly implies rhodamine B are largely loaded within the SiO2 particles (i.e., no more fluorescence quenching caused by locally concentrated rhodamine B within the SiO2 particles).
 | ||
| Fig. 3 SEM images and photos of rhodamine B dye loading within the mesoporous SiO2 particles and the subsequent surface modification of the SiO2 particles with MPS as well as the removal of the SiO2 particles corresponding absorption and fluorescent patterns. | ||
Fig. 4 presents the calibration curve of rhodamine B in an aqueous solution as a function of the concentration. The reliable instrumental response of the rhodamine B concentration was found to be from 0.04 μg mL−1 to 1.42 μg mL−1. The fluorescence intensities obtained at the concentrations above 1.42 μg mL−1 were not in the linear relationship due to the self-quenching behavior of rhodamine B at high concentrations (further increased concentrations exhibited decreased intensities).9,36 To quantify the amount of rhodamine within the mesoporous SiO2 particles, an aliquot of the dye-loaded particles after the extensive purification was then diluted with water and treated with a small quantity of HF to completely dissolve the SiO2 particles whose solution colors (the digital photos in Fig. 3) and fluorescence intensities were significantly increased. These observations clearly indicated that most dye molecules were encapsulated within the SiO2 particles. Although varying amounts of dye (e.g., 5.0–100.0 mg) were mixed with a fixed amount (i.e., 100.0 mg) of the SiO2 particles, the efficient loading of rhodamine B occurred at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of dye to SiO2 particles. Other ratios resulted in either the insufficient loading or inefficient loading of the dye within the SiO2 particles under our reaction conditions. For example, the use of higher ratios of dye to SiO2 particles required longer purification times and loaded a smaller fraction of the dye within the SiO2 particles; the use of lower ratios of dye to SiO2 particles (below 10 wt%) resulted in a much lower amount of dye loading than could be used in practical applications. Based on the 1:10 ratio of rhodamine B and the mesoporous SiO2 particles, the loading of the dye within the SiO2 particles was typically up to ∼4.5 wt% of the initial rhodamine B (i.e., ∼4.5 mg of rhodamine B in 100 mg of the SiO2 particles). These dye-loaded SiO2 particles were then diluted and served as cores to prepare temperature-responsive poly(NIPAM) layers via thermally- and light-induced radical polymerizations.
:10 ratio of dye to SiO2 particles. Other ratios resulted in either the insufficient loading or inefficient loading of the dye within the SiO2 particles under our reaction conditions. For example, the use of higher ratios of dye to SiO2 particles required longer purification times and loaded a smaller fraction of the dye within the SiO2 particles; the use of lower ratios of dye to SiO2 particles (below 10 wt%) resulted in a much lower amount of dye loading than could be used in practical applications. Based on the 1:10 ratio of rhodamine B and the mesoporous SiO2 particles, the loading of the dye within the SiO2 particles was typically up to ∼4.5 wt% of the initial rhodamine B (i.e., ∼4.5 mg of rhodamine B in 100 mg of the SiO2 particles). These dye-loaded SiO2 particles were then diluted and served as cores to prepare temperature-responsive poly(NIPAM) layers via thermally- and light-induced radical polymerizations.
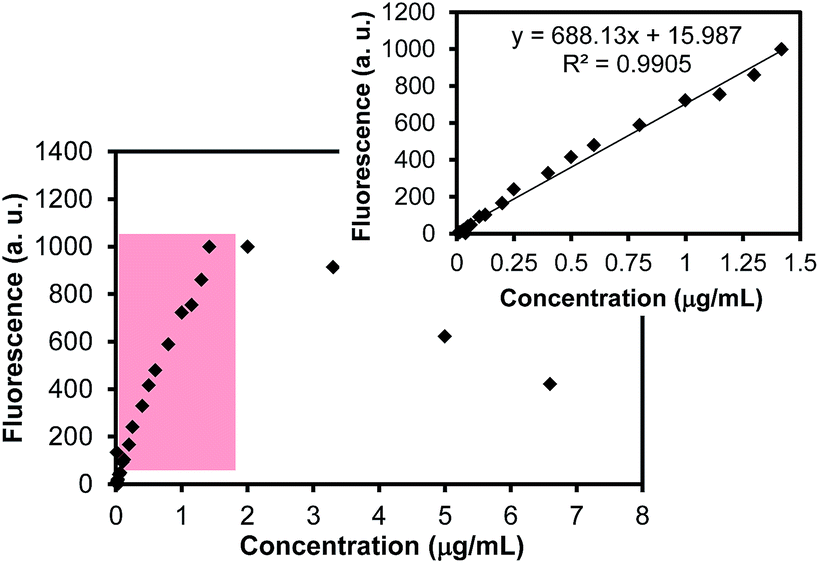 | ||
| Fig. 4 Fluorescence intensities of rhodamine B in aqueous solution as a function of concentration. The inset shows the reliable dynamic range (0.04 μg mL−1–1.42 μg mL−1) for quantitative analysis. | ||
The coating of poly(NIPAM) layers around the aforementioned MPS-modified SiO2 particles possessing rhodamine B was initially achieved in the presence of the APS initiator, NIPAM monomer, and BIS cross-linker via thermally-induced radical polymerization (Fig. 5). This polymerization method required a moderate reaction temperature of 70 °C for 6 hours to form cross-linked poly(NIPAM) shells around the dye-loaded SiO2 particles. The color of the solution developed from bright pink to very light milky-pink over the course of polymerization. After the completion of the reaction, the final solution was centrifuged to remove unreacted starting materials, oligomers, and leached rhodamine B. During this purification process, the color of the top solution was slight pink, indicating the leakage of the encapsulated rhodamine B from the SiO2 particles. In addition, the slow decolorization/decomposition of rhodamine B by the initiator APS was observed during the thermally-induced polymerization approach, which significantly lowered the total amount of loaded rhodamine B within the poly(NIPAM) particles (also responsible for the faded color of the solution). To confirm the decomposition process (it is known that azo-based dyes readily undergo decolorization/decomposition in the presence of persulfate),27 free rhodamine B and APS (similar to polymerization concentrations) in an aqueous solution were treated at 70 °C under argon and resulted in ∼100% loss within 2 hours, based on the fluorescence measurements. Similarly, the dye-loaded mesoporous SiO2 particles with APS were treated under 6 hours of the thermal polymerization conditions and still preserved about ∼30% of the initial amount of the loaded rhodamine B (i.e., exhibiting slow decomposition); the stability of rhodamine B is notably enhanced by encapsulating it in the SiO2 particles.36 As such, the polymer particles prepared by this approach still can possess the residual dye molecules after the removal of the SiO2 particles. Although rhodamine B is an interesting fluorescent marker, major leaching and decomposition (up to 70%) were inevitable throughout the thermally-induced polymerization method at 70 °C.
 | ||
| Fig. 5 Process of the preparation of a temperature-responsive poly(NIPAM) coating around the MPS-modified mesoporous SiO2 particles containing rhodamine B dye by thermally- and light-induced radical polymerization and the subsequent removal of the SiO2 particles along with SEM/TEM images (digital photos with * indicate unpurified solutions after polymerization). | ||
To reduce the significant dye-leaching and decomposition problems during polymerization, the formation of the poly(NIPAM) layers around the dye-loaded SiO2 particles was accomplished at a lower temperature in the presence of water soluble photo-initiators. Among well-known photo-initiators, including metal-complex-based compounds, benzophenone/thioxanthone/quinone derivatives, and organic acids,29–31,39,40 oxalic acid is one of the photoactive organic acids; it can effectively induce the polymerization of acrylamide and related monomers in an aqueous solution under light irradiation.29–31,33 We speculated that a strong sunlight source can induce a much more effective radical polymerization in the presence of these types of organic acid initiators than those of incandescent or fluorescent light sources, based on the previous studies.29,33 Thus, we employed oxalic acid as the photoinitiator for the formation of poly(NIPAM) layers around the dye-loaded SiO2 particles under the irradiation of a solar-simulated light (∼100 mW cm−2 of light intensity). Under our light-induced polymerization at room temperature, the color of the solution remained almost bright pink (i.e., there appeared to be no significant decolorization/decomposition of rhodamine B) as the polymerization progressed. The color of the final solution flask after being placed in an oven (above the LCST), however, was changed into slightly milky pink, apparently indicating the successful formation of poly(NIPAM). The final solution was then centrifuged to remove any unreacted starting materials, free poly(NIPAM), and leached rhodamine B. We noted that the color of the top solution after the centrifugation step was slightly pink, and this solution was subjected to a fluorescence measurement to evaluate the leached amount of the dye throughout the polymerization (∼30% loss by leaching). Separately, to examine the inevitable decolorization and/or decomposition of rhodamine B under the light-induced polymerization conditions, free rhodamine B and the dye-loaded mesoporous SiO2 particles were exposed to the solar-simulated light irradiation for 6 hours in the presence of oxalic acid, revealing a less than 15% and 7% loss of the initial amounts of rhodamine B, respectively. This clearly indicated that the azo-based compounds are stable under our light-induce polymerization conditions. We also speculated that the leaching of the guest molecules is relatively less favorable during the light-induced polymerization because of the reaction at room temperature, leading to much less decomposition. As such, this light-induced synthesis allowed for much less overall leaching and decomposition of the loaded dye during the formation of the poly(NIPAM) layers around the dye-loaded SiO2 particles, and this polymerization strategy could provide the additional advantage of possibly loading a wide range of thermally unstable guest molecules.
The SEM images show the size and distribution before and after the coating of the poly(NIPAM) layers around the dye-loaded mesoporous SiO2 particles prepared by thermally- and light-induced radical polymerizations (Fig. 5). The diameter of MPS-modified SiO2 particles loaded with rhodamine B was ∼450 nm while the polymer coated particles prepared by thermally- and light-induced radical polymerization were estimated to be ∼560 nm and ∼500 nm, respectively. The diameters of these particles were found to be much larger when measured by DLS (∼740 nm and ∼570 nm, respectively). This discrepancy is caused by the notable hydration and dehydration of the polymer shell layers around the SiO2 core particles in the aqueous solution (for the DLS measurements) and on a substrate under high vacuum conditions (for the SEM/TEM analyses). Upon the formation of the polymer layers, the PDI values of these poly(NIPAM)-coated mesoporous SiO2 particles prepared by both methods exhibited was much higher than that of the MPS-modified SiO2 particles due to the presence of the inter-particle assembly by the polymer shell layers and local agglomeration/aggregation of the particles in the solution. Overall, both polymerization approaches resulted in the reliable formation of temperature-responsive poly(NIPAM) layers around the SiO2 particles, but the polymer layers prepared by the light-induced approach resulted in a relatively thinner coating than those of the thermally-induced polymerization method under the same monomer and initiator ratios. More thorough studies to precisely control the thickness of the polymer layers around the SiO2 particles are underway.
The poly(NIPAM) coating around the dye-loaded SiO2 particles was subsequently treated with dilute hydrofluoric acid at a low temperature to selectively dissolve the SiO2 cores without disrupting the polymer layers, leaving the loaded rhodamine B within the poly(NIPAM) particles. The SEM and TEM images in Fig. 5 confirmed the formation of poly(NIPAM) layers around the SiO2 particles. In particular, the TEM images of the poly(NIPAM)-coated mesoporous SiO2 particles before and after treatment with the HF solution clearly showed the successful removal of the core particles. EDX analysis also confirmed the absence of the SiO2 particles after the HF treatment (data not shown), which was similar to our previous study.16 The diameters of poly(NIPAM) particles after the removal of the SiO2 cores largely decreased due to the full dehydration of poly(NIPAM) and the absence of the cores. The absence of the core particles also resulted in a significant decrease in the overall particle diameters and a noticeable structural change from a relatively smooth surface to an uneven surface (the diameters of these particles, however, were much larger in the DLS analysis due to the absence of the core and complete hydration of the particles in an aqueous solution, vide infra). For our drug-delivery applications, these two types of dye-loaded poly(NIPAM) particles prepared under the same concentration of the starting materials were then compared for their guest molecule capacity, temperature responsiveness, and triggered release properties below.
The hydrodynamic diameters of the dye-loaded poly(NIPAM) particles with and without SiO2 core particles were measured by DLS upon the heating and cooling of the solution (Fig. 6). When comparing the dye-loaded poly(NIPAM) particles prepared by the two polymerization methods, the initial diameter of the poly(NIPAM) particles prepared by thermally-induced polymerization is larger than that of the particles prepared by light-induced polymerization under the same reaction concentration; this indicates that there was a thicker coating of the poly(NIPAM) layers around the SiO2 particles, which was consistent with the previous SEM/TEM analyses. After the removal of the SiO2 cores, the diameters of the hollow-type poly(NIPAM) particles were estimated to be ∼780 nm (PDI: 0.38) for those that underwent thermally-induced polymerization and ∼680 nm (PDI: 0.34) for those that underwent light-induced polymerization well below the LCST (i.e., ≤25 °C). The diameters of these particles were found to be slightly larger than those particles possessing the SiO2 cores, presumably due to the absence of the core particles.4,16 Upon heating the particle solutions above 40 °C (i.e., well above the LCST of poly(NIPAM)), the diameters of the particles notably decreased from ∼780 to ∼325 nm (PDI: 0.30) and from ∼680 nm to ∼430 nm (PDI: 0.23), respectively. As the mesoporous SiO2 cores were removed, these poly(NIPAM) particles with the hollow-type structure caused much greater diameter changes, which may provide for a better triggered release of the loaded guest molecules. The distinctive volume transitions of poly(NIPAM)-based materials at physiological temperatures can be simply explained by the loss and gain of hydrogen bonds at the hydrophilic sites (–CO, –NH–) within the polymer particles.3,4,6,7,10,11,16,41 The loss of hydrogen bonding above the LCST of poly(NIPAM) causes the deswelling of the polymer particles and the recovery of the hydrogen bonds above the LCST results in the swelling of the polymer particles. These deswelling and swelling of poly(NIPAM)-based particles can offer precise triggered-release aspects in their potential drug-delivery applications.
 | ||
| Fig. 6 Reversible swelling and deswelling of the dye-loaded poly(NIPAM) and poly(NIPAM) on the dye-loaded SiO2 particles prepared by thermally- and light-induced radical polymerizations. | ||
To examine the thermally triggered release properties, aliquots of the samples (i.e., poly(NIPAM) particles containing rhodamine B after the removal of the SiO2 particles) prepared by the light- and thermally-induced polymerization were subjected to the fluorescence measurements at given time intervals during the dynamic dialysis at 40 °C (Fig. 7). As the poly(NIPAM) particles underwent significant volume reduction upon heating the solution well above the LCST (vide supra, DLS results), the quicker and greater release of the encapsulated rhodamine B was expected. The release of rhodamine B rapidly increased at the beginning of the dialysis hours (≤10 h); the poly(NIPAM) particles prepared by the light-induced polymerization exhibited faster and greater release than their counterparts, probably due to the thinly-coated polymer shells. As these poly(NIPAM) particles were prepared in the presence of over 7 mol% of the BIS cross-linker, the thick polymer layers prepared by thermally-induced polymerization notably prevented the full release of the encapsulated rhodamine B. This was also confirmed by the residual pink aggregates found in a dialysis bag that could be seen even after 1 week of dialysis, where the thin polymer particles prepared by the light-induced method did not exhibit visible pink aggregates. The total release of rhodamine B was estimated to be about 45% for the particles prepared by thermally-induced polymerization and about 80% for the particles prepared by light-induced polymerization. It is also important to remember that the initial total amounts of loaded rhodamine B within the poly(NIPAM) particles were noticeably different due to the decolorization/decomposition and leaching of the loaded rhodamine B under the two different radical polymerization regimes. As control experiments, the release of the dye-loaded poly(NIPAM) particles prepared by both methods was monitored when these samples were kept well below the LCST (i.e., ≤25 °C). These poly(NIPAM) particles showed a relatively slow release of rhodamine B, which was mainly caused by diffusion through the pores of the polymer layers (data not shown). The dye-loaded mesoporous SiO2 particles coated with poly(NIPAM) layers above the LCST also exhibited a slow release of rhodamine B; this occurred because as the poly(NIPAM) layers moderately underwent deswelling, the mesoporous SiO2 particles possessing rhodamine B as a core prevented the temperature-induced collapse of the particles to fully release the loaded rhodamine B. Based on these fluorescence measurements, the solar-simulated light-induced radical polymerization readily allowed for much less leakage and decomposition of rhodamine B within the poly(NIPAM) particles and a slightly quick response to an external stimulus (i.e., upon heating the solution temperature above 40 °C). These overall results are comparable to the controlled-release properties of poly(NIPAM)-based materials,15,42,43 which were well supported by their precisely tunable swelling and deswelling behavior above and below the LCST (Fig. 6).
 | ||
| Fig. 7 Rhodamine B release profiles from the dye-loaded poly(NIPAM) particles above the LCST (i.e., 40 °C) prepared by thermally- and light-induced radical polymerizations. | ||
As a whole, our results in this stage clearly demonstrated that poly(NIPAM) particles prepared by the light-induced approach at room temperature allowed for the retention of a higher concentration of guest molecules and a quicker triggered-release responsiveness than those prepared by the thermally-induced method. Further studies will involve the generalization of our strategy to design stimuli-responsive polymer particles exhibiting the efficient loading of a wide range of chemotherapeutic drugs (e.g., Withaferin A, Cisplatin, and Taxol) and the tunably controlled release of the encapsulated drugs for the development of novel drug-delivery and treatment systems.
Conclusions
Our simple strategies showed that the temperature-responsive poly(NIPAM) layer around the guest molecule-loaded mesoporous SiO2 particle was reliably formed and demonstrated the particles' loading capacity of rhodamine B. SEM/TEM revealed the formation of the poly(NIPAM) layer on the dye-loaded SiO2 particles via both thermally- and light-induced radical polymerizations, as well as the successful removal of the sacrificial SiO2 core template with dilute hydrofluoric acid, which left the loaded dye molecules behind within the polymer particles. Fluorescence and DLS measurements proved that the particles prepared by the light-induced approach allowed for significantly less leaching and decolorization/decomposition of the encapsulated dye during polymerization and the formation of the relatively thin coating of the poly(NIPAM) layer than those samples prepared by thermally-induced polymerization. In addition, these relatively thin poly(NIPAM) particles underwent a quicker controlled release of the loaded guest molecules. The development of biocompatible polymer-based delivery vehicles possessing a high dose of interesting guest molecules and exhibiting external stimuli responsiveness can lead to a variety of promising applications in the fields of biomedical imaging and therapy, because these types of carriers may be able to minimize the negative side effects associated with delivery-based treatments.Acknowledgements
We gratefully acknowledge the financial support from the Cottrell College Science Award of Research Corporation and Illinois State University. This research is also supported by the Korea Ministry of Environment as “Global Top Project” (Project no: GT-14-B-01-002-0) and the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science, and Technology (no. 2012R1A1A4A01019566). We would also like to acknowledge the support from the Synchrotron Light Research Institute (a public organization) and the Graduate School and Faculty of Science at Chiang Mai University in Thailand.References
- T. A. Allen and P. C. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- R. Atkin, M. Bradley and B. Vincent, Soft Matter, 2005, 1, 160–165 RSC.
- Z. Dai and T. Ngai, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2995–3003 CrossRef CAS PubMed.
- H. Gao, W. Yang, K. Min, L. Zha, C. Wang and S. Fu, Polymer, 2005, 46, 1087–1093 CrossRef CAS PubMed.
- C. M. Nolan, L. T. Gelbaum and L. A. Lyon, Biomacromolecules, 2006, 7, 2918–2922 CrossRef CAS PubMed.
- S. R. Sershen, S. L. Westscott and N. J. Halas, J. Biomed. Mater. Res., 2000, 51, 293–298 CrossRef CAS.
- L. E. Strong and J. L. West, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 307–317 CrossRef CAS PubMed.
- Y. Zhang and A. L. Yarin, J. Mater. Chem., 2009, 19, 4732–4739 RSC.
- S.-M. Lee, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2010, 132, 17130–17138 CrossRef CAS PubMed.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- S.-M. Lee, H. Chen, C. M. Dettmer, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2007, 129, 15096–15097 CrossRef CAS PubMed.
- C. Liu, J. Guo, W. Yang, J. Hu, C. Wang and S. Fu, J. Mater. Chem., 2009, 19, 4764–4770 RSC.
- S. R. MacEwan, D. J. Callahan and A. Chiloti, Nanomedicine, 2010, 5, 793–806 CrossRef CAS PubMed.
- J. Qian and F. Wu, J. Mater. Chem. B, 2013, 1, 3464–3469 RSC.
- N. Singh, A. Karambelkar, L. Gu, K. Lin, J. S. Miller, C. S. Chen, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2011, 133, 19582–19585 CrossRef CAS PubMed.
- J.-H. Kim, R. D. Burnett and A. Gabriel, J. Biomed. Nanotechnol., 2012, 8, 432–438 CrossRef CAS PubMed.
- L. Sun, X. Zhang, C. Zheng, Z. Wu, X. Xia and C. Li, RSC Adv., 2012, 2, 9904–9913 RSC.
- J. Zeng, P. Du and P. Liu, RSC Adv., 2013, 3, 19492–19500 RSC.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, C. P. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- J. Tu, N. Li, Y. Chi, S. Qu, C. Wang, Q. Yuana, X. Li and S. Qiu, Mater. Chem. Phys., 2009, 118, 273–276 CrossRef CAS PubMed.
- L. Yan, Z.-K. Wang, J.-J. Yan, L.-F. Han, Q.-H. Zhou and Y.-Z. You, Polym. Chem., 2013, 4, 1243–1249 RSC.
- Y. Li, Y. Wu, C. Luo, F. Yang, L. Qin, T. Fu, G. Wei, X. Kang and D. Wu, J. Mater. Chem. B, 2013, 1, 4644–4654 RSC.
- L. Sun, X. Zhang, Z. Wu, C. Zheng and C. Li, Polym. Chem., 2014, 5, 1999–2009 RSC.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
- X. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940–7945 CrossRef CAS PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- Y. Ding, L. Zhu, J. Yan, Q. Xiang and H. Tang, J. Environ. Monit., 2011, 13, 3057–3063 RSC.
- A. H. Mcheik and M. M. El Jamal, J. Chem. Technol. Metall., 2013, 48, 357–365 CAS.
- R. Gangopadhyay and P. Ghosh, Eur. Polym. J., 2000, 36, 1579–1606 CrossRef.
- P. Ghosh and R. Gangopadhyay, Eur. Polym. J., 2000, 36, 625–634 CrossRef CAS.
- M. A. Tasdelen, B. Karagoz, N. Bicak and Y. Yagci, Polym. Bull., 2008, 59, 759–766 CrossRef CAS.
- P. Weiss, Pure Appl. Chem., 1967, 15, 587–600 CrossRef CAS.
- H. J. L. Bressers and J. G. Kloosterboer, Polym. Bull., 1980, 2, 201–204 CrossRef CAS.
- R. Gui and H. Jin, RSC Adv., 2014, 4, 2797–2806 RSC.
- J. Y. Bae, O.-H. Park, J.-I. Jung, K. T. Ranjit and B.-S. Bae, Microporous Mesoporous Mater., 2004, 67, 265–271 CrossRef CAS PubMed.
- Z. Gan, J. Ju, T. Zhang and D. Wu, J. Nanomater., 2011, 2011, 8, DOI:10.1155/2011/753705.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Oison, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- M. Ratanajanchai, D. Tanwilai and P. Sunintaboon, J. Colloid Interface Sci., 2013, 409, 25–31 CrossRef CAS PubMed.
- C. R. Rivarola, M. A. Biasutti and C. A. Barbero, Polymer, 2009, 50, 3145–3152 CrossRef CAS PubMed.
- J.-H. Kim and T. R. Lee, Chem. Mater., 2004, 16, 3647–3651 CrossRef CAS.
- L. E. Strong, S. N. Dahotre and J. L. West, J. Controlled Release, 2014, 178, 63–68 CrossRef CAS PubMed.
- B. M. Teo, S. W. Prescott, G. J. Price, F. Grieser and M. Ashokkumar, J. Phys. Chem. B, 2010, 114, 3178–3184 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |