
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of high molecular weight polyglycolide in supercritical carbon dioxide†
Christian Schmidtac,
Marc Behlb,
Andreas Lendlein*ab and
Sabine Beuermann*ac
aInstitute of Chemistry, University of Potsdam, Karl-Liebknecht Str. 24-25, 14476 Potsdam, Germany
bInstitute of Biomaterial Science, Helmholtz-Zentrum Geesthacht, Kantstraße 55, 14513 Teltow, Germany
cInstitute of Technical Chemistry, Clausthal University of Technology, Arnold-Sommerfeld-Str. 4, 38678 Clausthal-Zellerfeld, Germany
First published on 29th July 2014
Abstract
Polyglycolide (PGA) is a biodegradable polymer with multiple applications in the medical sector. Here the synthesis of high molecular weight polyglycolide by ring-opening polymerization of diglycolide is reported. For the first time stabilizer free supercritical carbon dioxide (scCO2) was used as a reaction medium. scCO2 allowed for a reduction in reaction temperature compared to conventional processes. Together with the lowering of monomer concentration and consequently reduced heat generation compared to bulk reactions thermal decomposition of the product occurring already during polymerization is strongly reduced. The reaction temperatures and pressures were varied between 120 and 150 °C and 145 to 1400 bar. Tin(II) ethyl hexanoate and 1-dodecanol were used as catalyst and initiator, respectively. The highest number average molecular weight of 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 was obtained in 5 hours from polymerization at 120 °C and 530 bar. In all cases the products were obtained as a dry white powder. Remarkably, independent of molecular weight the melting temperatures were always at (219 ± 2) °C.
200 g mol−1 was obtained in 5 hours from polymerization at 120 °C and 530 bar. In all cases the products were obtained as a dry white powder. Remarkably, independent of molecular weight the melting temperatures were always at (219 ± 2) °C.
1. Introduction
Polyglycolide (PGA) is of high interest for medical applications such as implants or drug release systems.1–4 Compared to other dilactone based polyesters PGA degrades fast. Therefore a relatively high molecular weight is required to guarantee a sufficient residence time of the PGA for the therapy. However, the thermal decomposition of PGA starts already below the melting transition (Tm) of the PGA. When diglycolide is polymerized in the melt, the Tms of the oligo- and polyglycolide increase rapidly with increasing molecular weights. Accordingly the reaction temperature needs to be raised as well to avoid solidification of the reaction mixture. Here it needs to be considered that the polymerization of diglycolide is a strong exothermic reaction. An additional challenge to be met is the processing of the PGA into the intended application relevant bodies. The solubility of PGA in common solvents, which are suitable for biomaterials, is low and can only be achieved by processing at low temperatures at high pressures. Otherwise thermal degradations occurs, which becomes apparent by a brown coloring of the PGA.Supercritical carbon dioxide (scCO2) possesses several favorable characteristics as reaction medium for polymer synthesis. It is a chemically inert, non-toxic, and environmentally benign gas, which can be easily separated from the product by reducing the pressure.5–7 Further, CO2 is well soluble in most polymers resulting in polymer swelling, which changes the mechanical and physical properties of the material. Therefore, CO2 can reduce the melting temperature (Tm) and the glass transition temperature (Tg) of polymers and monomers. The latter is caused by the plasticizing action of CO2.5 For example, the Tg of poly(methyl methacrylate) (PMMA) is lowered by up to 60 °C at 100 bar in a CO2 atmosphere.5,8,9 Supercritical carbon dioxide was used as reaction medium for a wide variety of monomers encompassing conventional (meth)acrylates, fluoroolefins, and cyclic monomers leading to biodegradable polymers.10–22
It was hypothesized that scCO2 could be used as reaction medium to keep the PGA soluble during polymerization and by this avoid high reaction temperatures whereby high Mn can be achieved. In addition, scCO2 has proven to be an excellent process medium for particle formation.23,24 Since particles of well-defined size are essential for the controlled drug delivery a combined process combing synthesis and shaping appears to be highly attractive.
Previously, ring-opening polymerizations (ROP) of cyclic monomers were carried out in scCO2 as reaction medium using tin(II) ethyl hexanoate (SnEH2) as catalyst.19,22 The polymerization mechanism for the cyclic diester diglycolide as monomer following a coordination-insertion mechanism. First, SnEH2 reacts with dodecanol to form the catalytic active tin(II) alkoxide. The alkoxide coordinates monomer and successive ring-opening of the cyclic monomer occurs. Then, additional monomer units are coordinated and inserted between tin and the previously added monomer unit leading to chain growth. The polymerization is terminated by adding an excess amount of ethyl acetate, leaving an acetyl terminal group in the polymer.
The goal of this work was to explore whether high molecular weight PGA may be obtained using supercritical carbon dioxide as reaction medium and SnEH2 as catalyst. Since no information are available for ROP of diglycolide in non-stabilized systems under high-pressure conditions several monomer and catalyst concentrations as well as pressures and temperatures were explored.
2. Experimental section
Materials
Diglycolide (purity 99.9%, Sigma, Munich, Germany) was recrystallized from a minimum amount of dry toluene. Tin(II) ethyl hexanoate (purity 95%, Sigma), 1-dodecanol (purity 99.8% Fluka), toluene (purity 99.9%, Sigma), ethyl acetate (purity 99%, VWR) were used as received. CO2 (purity 99.998%) was obtained from Air Liquide (Düsseldorf, Germany).Typical polymerization procedure
PGA synthesis under scCO2 conditions was performed in an optical high-pressure cell (RGT 601, material no. 2.4668, Arbed Saarstahl) with an inner volume of 6 mL. A detailed description is given in ref. 25. The cell was closed on one side and purged with argon before diglycolide (500 mg, 4.4 × 10−3 mol) as well as a solution of tin(II) ethyl hexanoate (0.05 mg, 1.2 × 10−7 mol) and 1-dodecanol (0.18 mg, 9.7 × 10−7 mol) in toluene (0.1 mL) were quickly transferred into the cell. Afterwards the cell was sealed, CO2 was added and the required pressure was adjusted using a manual high-pressure generator. To control temperature a resistive heating element (CGE Asthom) and a PID-controller (Eurotherm 815) were used. The reaction was quenched by releasing the pressure followed by quickly opening the cell to allow for fast removal of the product. The product is obtained as a white powder, as shown in Fig. 1. The crude product was isolated and subsequently purified by Soxhlet extraction with ethyl acetate for 4 hours, which resulted in a white powder. Monomer conversions were determined gravimetrically. | ||
| Fig. 1 PGA synthesized using scCO2 at 120 °C (sample 30). | ||
Characterization
| Sample | T [°C] | p [bar] | t [min] | cM [mol L−1] | ccat [mol L−1] | cI [mol L−1] | nM/ncat/nI | x [%] | Mn [g mol−1] | PDI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 130 | 500 | 60 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
55 | 3900 | 1.3 |
| 2 | 130 | 500 | 120 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
70 | 4000 | 1.4 |
| 3* | 130 | 600 | 300 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
22 | 16500 |
1.7 |
| 4* | 130 | 590 | 840 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
48 | 22500 |
1.2 |
| 5* | 150 | 600 | 60 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
60 | 14600 |
2.2 |
| 6* | 150 | 570 | 120 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
74 | 14000 |
1.4 |
| 7* | 150 | 560 | 180 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
95 | 30000 |
1.9 |
| 8* | 150 | 840 | 300 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
55 | 17400 |
1.3 |
| 9 | 150 | 540 | 840 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
91 | 8900 | 1.6 |
| 10 | 120 | 550 | 300 | 0.72 | 2.06 × 10−5 | 1.61 × 10−4 | 35000/1/7.9 |
23 | 14100 |
1.5 |
| 11 | 120 | 510 | 300 | 1.44 | 4.12 × 10−5 | 3.22 × 10−4 | 35000/1/7.9 |
18 | 22800 |
1.4 |
| 12 | 120 | 530 | 300 | 2.88 | 8.24 × 10−5 | 6.44 × 10−4 | 35000/1/7.9 |
13 | 31200 |
1.3 |
| 13 | 120 | 800 | 300 | 0.72 | 1.23 × 10−5 | 1.61 × 10−4 | 58000/1/13 |
50 | 3000 | 1.4 |
| 14 | 120 | 530 | 300 | 0.72 | 1.65 × 10−5 | 1.61 × 10−4 | 44000/1/9.8 |
42 | 3200 | 1.3 |
| 15 | 120 | 400 | 300 | 0.72 | 1.85 × 10−5 | 1.61 × 10−4 | 39000/1/8.7 |
17 | 5900 | 1.3 |
| 16 | 120 | 660 | 300 | 0.72 | 2.06 × 10−5 | 1.61 × 10−4 | 35000/1/7.9 |
18 | 10700 |
1.8 |
| 17 | 120 | 880 | 300 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
40 | 4100 | 2.1 |
| 18 | 120 | 460 | 840 | 0.72 | 1.65 × 10−5 | 1.61 × 10−4 | 44000/1/9.8 |
65 | 4500 | 1.6 |
| 19 | 120 | 210 | 840 | 0.72 | 2.06 × 10−5 | 1.61 × 10−4 | 35000/1/7.9 |
34 | 5900 | 1.3 |
| 20* | 120 | 430 | 840 | 0.72 | 2.67 × 10−5 | 1.61 × 10−4 | 27000/1/6 |
34 | 8200 | 1.6 |
| 21 | 120 | 430 | 840 | 0.72 | 3.09 × 10−5 | 1.61 × 10−4 | 23000/1/5.2 |
61 | 4900 | 1.6 |
| 22 | 120 | 210 | 840 | 0.72 | 2.67 × 10−5 | 8.05 × 10−5 | 27000/1/3 |
79 | 5900 | 1.3 |
| 23* | 120 | 200 | 840 | 0.72 | 2.67 × 10−5 | 1.61 × 10−4 | 27000/1/6 |
21 | 14600 |
1.8 |
| 24* | 120 | 145 | 840 | 1.44 | 5.34 × 10−5 | 1.61 × 10−4 | 27000/1/3 |
24 | 11100 |
1.4 |
| 25 | 120 | 480 | 840 | 1.44 | 5.34 × 10−5 | 1.61 × 10−4 | 27000/1/3 |
21 | 13000 |
1.4 |
| 26 | 120 | 430 | 300 | 1.08 | 3.09 × 10−5 | 2.42 × 10−4 | 35000/1/7.9 |
24 | 15100 |
1.4 |
| 27 | 120 | 1030 | 300 | 1.08 | 3.09 × 10−5 | 2.42 × 10−4 | 35000/1/7.9 |
12 | 8400 | 1.4 |
| 28 | 120 | 1250 | 300 | 1.08 | 3.09 × 10−5 | 2.42 × 10−4 | 35000/1/7.9 |
9 | 10800 |
1.5 |
| 29 | 120 | 1410 | 840 | 1.08 | 3.09 × 10−5 | 2.42 × 10−4 | 35000/1/7.9 |
41 | 3900 | 1.6 |
| 30 | 120 | 360 | 840 | 1.44 | 4.12 × 10−5 | 3.22 × 10−4 | 35000/1/7.9 |
22 | 23100 |
1.8 |
3. Results and discussion
The polymerization conditions and the experimental results are summarized in Table 1. The polymerization kinetics were explored by quenching the reactions after different reaction times t and determining the number average molecular weight Mn, whereby the other reaction conditions were kept identical. As shown in Fig. 2 an increase in t at 130 °C resulted in an increase in Mn to 22500 g mol−1 after 14 h (samples 1 to 4). When T was raised to 150 °C a PGA with Mn of 30000 g mol−1 was obtained after 3 h (sample 7). It should be noted that only a fraction of the polymer material was soluble in HFIP and analyzed via SEC. Thus, the true Mn value is expected to be even higher. A further increase in t resulted in a material that is completely soluble in HFIP and exhibits a decrease in Mn (Fig. 2, samples 5 to 9 in Table 1), which might be attributed to thermal decomposition. It is remarkable to note that even at 150 °C the PGA was obtained as a white powder. At first sight the finding of white polymer might be in conflict with the decrease in Mn at higher reaction times. However, low molecular weight decomposition products could be extracted by scCO2 during the depressurization process. The data indicate that an increase in T results in an enhancement of molecular weight and conversion. Because of the complex interplay between polymerization conditions and thermal decomposition of the polymer, the reaction conditions were varied with respect to cM, ccat, p, and T.
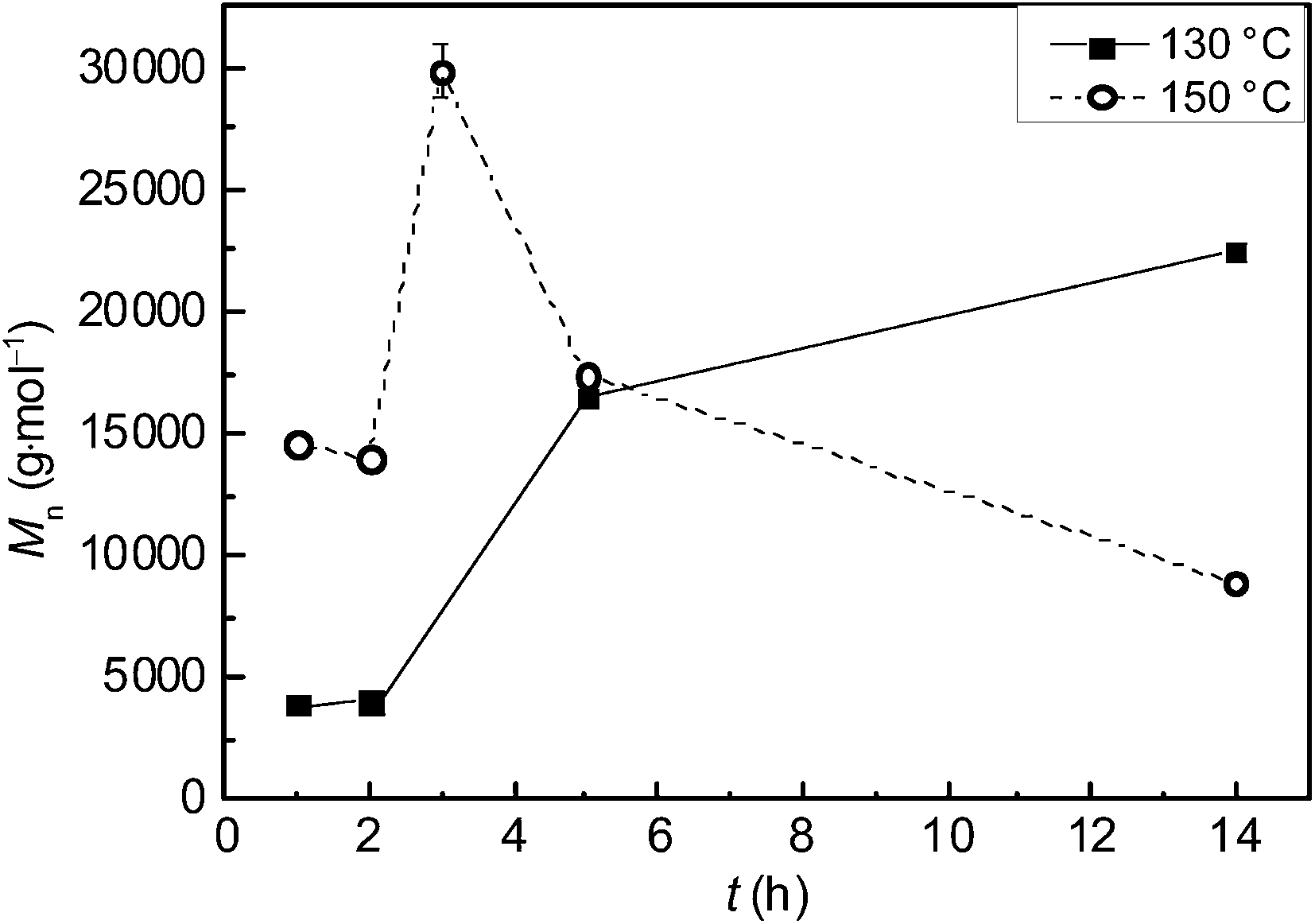 | ||
| Fig. 2 Influence of reaction time t on the number average molecular weight Mn of PGA polymerized in the presence of scCO2 at ■ 130 °C and ○ 150 °C. The lines indicate guides for the eye, margin of error are in most cases smaller than symbol size. | ||
Consideration of the monomer concentration shows that when cM was raised from 0.72 (sample 10) to 2.88 mol L−1 (sample 12) Mn increased from 14100 to 31200 g mol−1 at 120 °C. The corresponding molecular weight distributions are shown in Fig. 3 A. Sample 12 obtained with cM = 2.88 mol L−1 shows the highest Mn that was obtained within this study. Further, the molecular weight distributions do not show significant amounts of low molecular weight material. At first sight, this finding is surprising, since frequently ring-opening polymerizations yield cyclic oligomers as by-products. On the other hand, a recent kinetic study on ROP of glycidol revealed that side reactions being important at ambient pressure polymerizations are completely eliminated at elevated pressure.26 1H-NMR spectra of PGA were recorded in a mixture of HFIP and CDCl3. In addition to the solvent-derived peaks, only a single peak at 4.87 ppm is observed, which is assigned to CH2 in the polymer. This finding also suggests that significant amounts of oligomeric side products were not obtained. A typical 1H-NMR spectrum and a description of the sample preparation are contained in the ESI.†
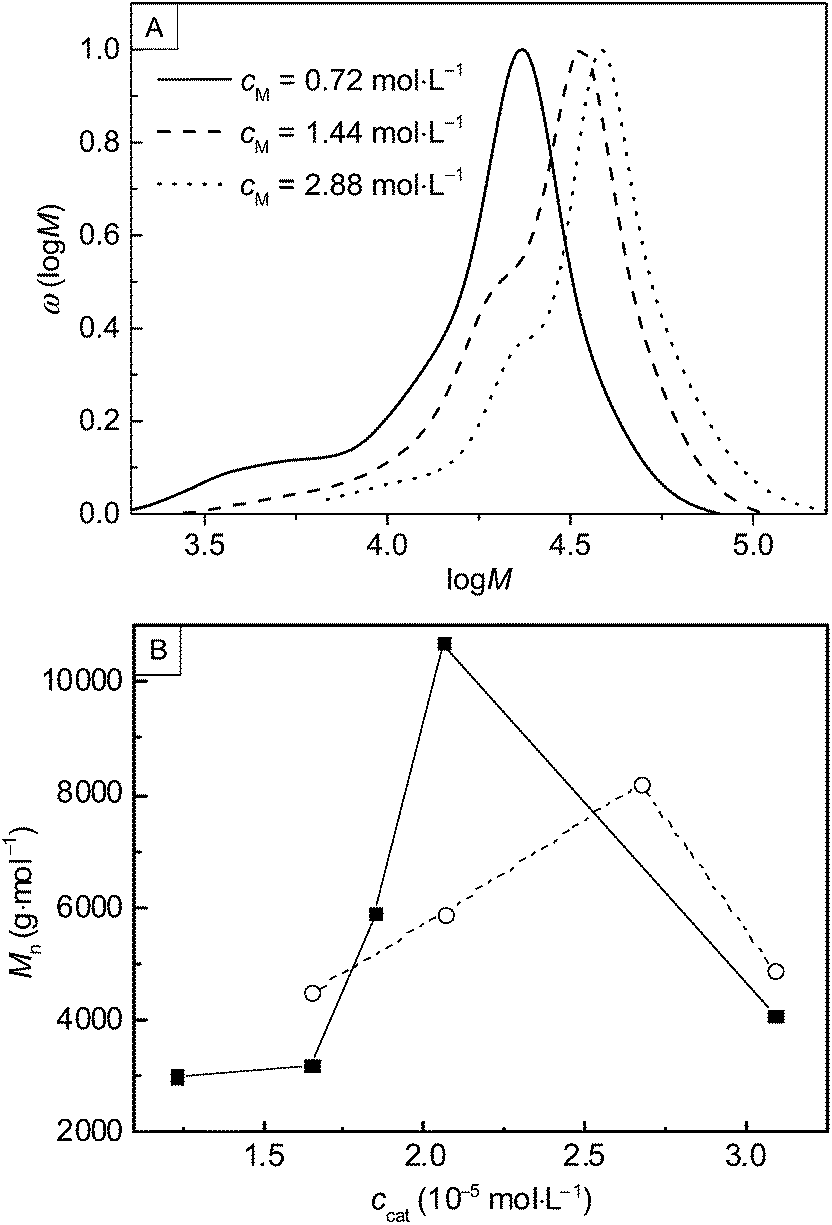 | ||
| Fig. 3 (A) Molecular weight distributions of PGA obtained from reactions with varying cM at 120 °C, 500 bar, and 5 hours with nM/ncat/nI = 35000/1/7.9: (I) 0.72 mol L−1 (II) 1.44 mol L−1 (III) 2.88 mol L−1. (B) Number average molecular weights Mn as a function of ccat at (○) 120 °C, 660 bar, 5 hours, cM = 0.72 mol L−1 and (■) at 120 °C, 400 bar, 14 hours, cM = 0.72 mol L−1. | ||
In addition, the influence of the concentrations of catalyst, ccat, on the molecular weight has been investigated. The results obtained for the variation of ccat are presented in Fig. 3B (samples 13 to 17 and 18 to 21). The experiments suggest that ccat has an optimum value to yield high molecular weight PGA. However, conversion seems to be minimal for these values. A similar behaviour is found for the variation of the dodecanol concentration, cI:Mn increases from 5900 to 14600 g mol−1 upon raising cI from 8.05 × 10−5 to 1.61 × 10−4 mol L−1 (samples 22 and 23), while conversions decrease from 79 to 21%. It is highly likely that these observations may be attributed to the influence of water. Dodecanol acts as a protic co-initiator to increase the nucleophilicity of the catalyst and enables the ring-opening reaction.2,27 Water fulfils the same task as dodecanol, but leads to shorter chain lengths. Therefore, it is required to either use completely dried materials or to increase cI to suppress the activation of the catalyst by water molecules. As sample 23 shows, by enhancing cI the activation of catalyst molecules by the initiator is promoted yielding polymer with higher Mn.
The influence of ccat can be explained by a delicate interplay as an increasing number of catalyst molecules at constant cI favour the activation by water molecules. However, the data suggests that the effect occurs also at low ccat. For low ccat low Mn at increased conversion were determined. It is most likely, that the increased relative concentration of aqueous impurities compared to active species causes an enhancement of the catalyst activation by water.
The data in Table 1 suggests that the molecular weight is also influenced by pressure (p). When p was increased from 145 (sample 24) to 480 bar (sample 25) Mn increased from 11100 to 13000 g mol−1. Such a behaviour was expected as at low pressures significant changes of the CO2 properties occur when p is increased. Once a certain pressure level is reached the physico-chemical properties change only slightly,5 and only a small influence is expected to occur. On the contrary, the molecular weight distributions shown in Fig. 4 indicate that an increase in pressure from 430 to 1250 bar (sample 26 to 28) leads to a distinct decrease in the peak molecular weight of the distribution while dispersities are rather similar. Moreover, with increasing pressure monomer conversions are significantly lowered.
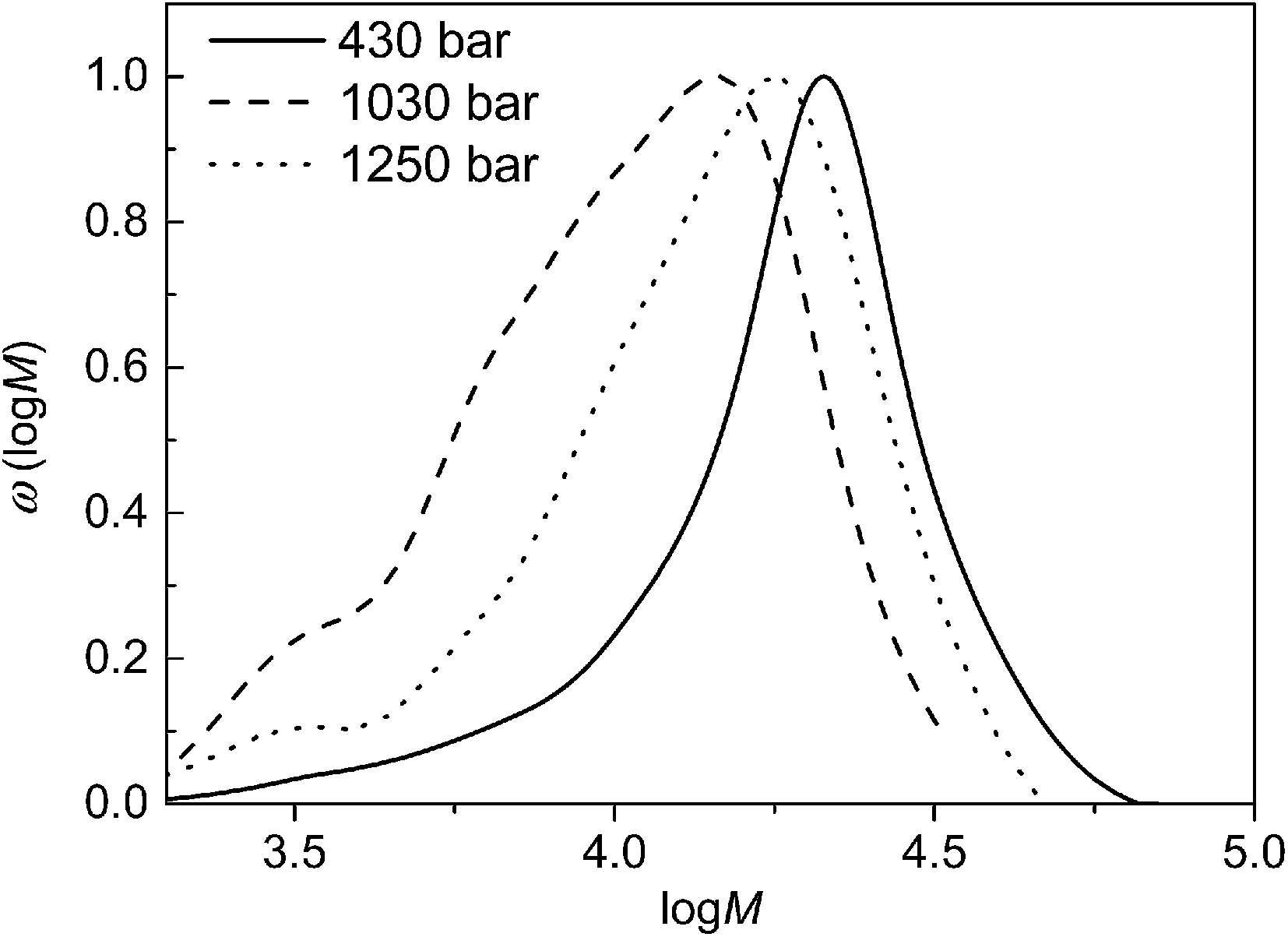 | ||
| Fig. 4 Molecular weight distributions obtained at the indicated pressures and the following reaction conditions: 5 hours, 120 °C, cM = 1.08 mol L−1, nM/ncat/nI = 35000/1/7.9. | ||
The pressure influence on the molecular weight distribution may be explained as follows. Firstly, the polymerization mechanism is considered. The ring-opening of the cyclic diester results in an increase in the molar volume, and thereby, this step is hindered at high pressure. This reasoning is in agreement with the finding that a reaction leading to a cyclic or a linear product might be directed towards the cyclic product at low pressure, whereas the linear product is obtained at high pressure.28 Moreover, interactions of the catalyst with CO2 need to be considered. Previously, it was shown that ROP in scCO2 with Sn catalysts proceeds slower at higher pressure. It was discussed that CO2 competes with the coordination of the monomer at the catalyst site, thus, reducing the coordination of the monomer.17,18 The coordination of the catalyst with CO2 was suggested to be reversible since no carbonate species were obtained after the polymerization. In addition, it was reported that the effect is less pronounced at higher temperature. While the literature mostly refers to polymerizations at 40 °C in this work the lowest temperature is 120 °C. Thus, it is anticipated that the coordination with CO2 is much less important. This reasoning is supported by the following finding:19 it was shown that at otherwise identical conditions ROP of ε-caprolactone at 80 °C in toluene proceeds only by a factor of 1.3 faster than in scCO2. The moderate decrease in the presence of scCO2 was explained with a fast equilibrium and the rather short time interval in which the chain ends are in the dormant carbonated state.
On the other hand the linear monomer units in the polymer chains might be more closely packed, which should lead to a higher polymerization rate at higher pressure. In addition, reaction rates are often increased at higher pressure due to higher monomer densities.5,29 Another important aspect is the phase behaviour of the monomer–polymer–CO2 system. Generally, with increasing pressure the solubility of polymer and monomer in CO2 is improved.5,29,30 Because of the opposing influences of pressure on the polymerization the reaction pressure should be at an intermediate level to yield high molecular weight polymer.
These observations show that there is a complex interplay between the relevant reaction parameters. Therefore, in future it is crucial to determine the optimum reaction conditions to yield high molecular weight polymer. In this study PGA with the highest number average molecular weight of 31200 g mol−1 was obtained after 5 h at 120 °C with cM = 2.88 mol L−1 and nM/ncat/nI of 35000 to 1 to 7.9 at 530 bar (sample 12). Since the reaction conditions were not yet optimized with respect to the presence of water, it is anticipated that even higher molecular weight material may become available.
To the best of our knowledge previously only Bratton, Brown and Howdle reported on the synthesis of PGA with scCO2 as reaction medium.22 The reactions were carried out for 24 hours in dispersion using a hydrocarbon stabilizer at 80 °C and pressures around 250 bar. As catalyst system SnEH2 and butanol were used. In all cases Mn values ranging from 4200 to 4900 g mol−1 were derived. These values are significantly lower than obtained in our work. The reasons may be seen in the lower temperature and the dispersed nature of the system in ref. 22. During the suspension polymerization the CO2-induced lowering of Tm should not play an important role.
The molecular weights of the PGA samples were determined by SEC analyses. In addition, MALDI-TOF was used to characterize some of the polymer samples. The MALDI-TOF mass spectrum of a sample with an SEC-derived Mn of 3900 g mol−1 (full distribution given in Fig. 5A) shows a molecule ion peak at m/z = 5420 g mol−1 (Fig. 5B) (sample 29), which correlates with 89 monomer units, the end groups 1-dodecanol and acetyl originating from the catalyst and termination with ethyl acetate, respectively, and a Na+ ion. In the MALDI-TOF spectrum the signals show a characteristic difference m/z = 116 g mol−1, which can be attributed to a diglycolide unit. Signals separated by m/z = 58 g mol−1, reflecting the mass of a glycolide unit, were not found. The molecular weight distribution and the MALDI-TOF mass spectra in Fig. 5 show some deviations in shape and the molecular weights are different. Jackson et al. showed that MALDI-TOF mass spectra are not representative for the molecular weight distribution in cases where the dispersities are higher than 1.2.31 That effect is due to instrumental limitations of MALDI-TOF and the use of different scaling of the data by both methods. The differences in Mp (most probable peak) values determined by SEC and MALDI-TOF are strongly increased for higher PDI.31,32 As the measured sample shows a SEC-derived PDI of 1.6 such a behaviour may be expected. In addition, differences in molecular weight may be caused by the PMMA calibration of the SEC set-up.
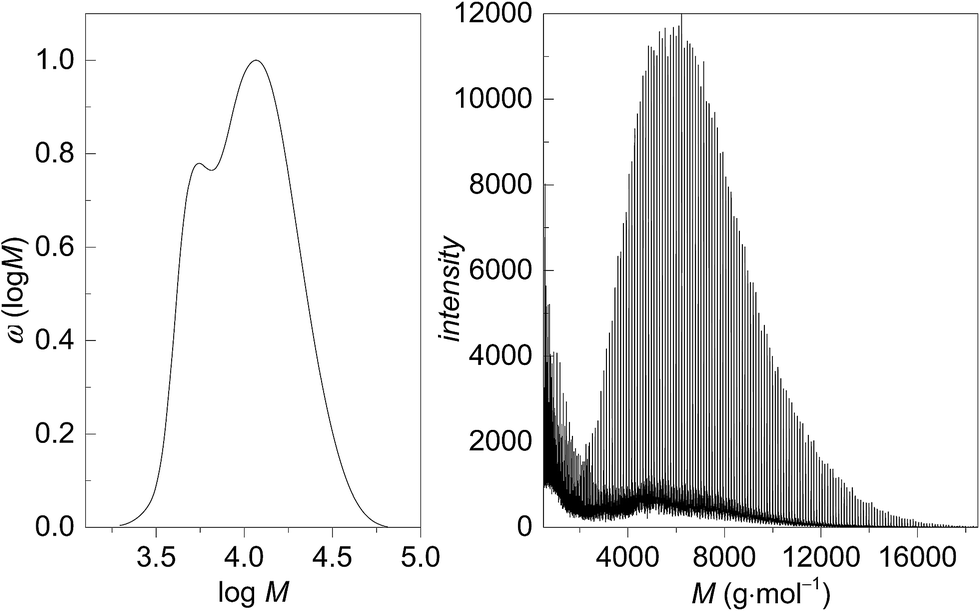 | ||
| Fig. 5 Comparison of (A) SEC-derived molecular weight distribution and (B) MALDI-TOF mass spectrum. | ||
FT-IR spectra allowed for an easy identification of the polymer. The spectra given in Fig. 6 clearly show that most bands are present in both polymer and monomer spectra. However, in the wavenumber range from 1600 to 500 cm−1 the monomer related absorption bands show a strongly different shape compared to the polymer spectra. Particularly important is the area around 1250 cm−1, which is assigned to the OCO stretching vibration. The change of the absorption behaviour is most likely a result of the transformation of the cyclic monomer to a linear polymer. These absorption bands are highly specific for the polymer and independent of its Mn indicating the synthesis of polyglycolide in all cases. Elemental analyses confirm these conclusions. The data shows just slight variations in the polymer's elemental contents. The carbon content varies between 40.5 and 41.4% (Ccalculated = 41.4%), the hydrogen content between 2.5 and 2.7% (Hcalculated = 3.4%) and the oxygen content between 52.9 and 53.5% (Ocalculated = 55.2%).
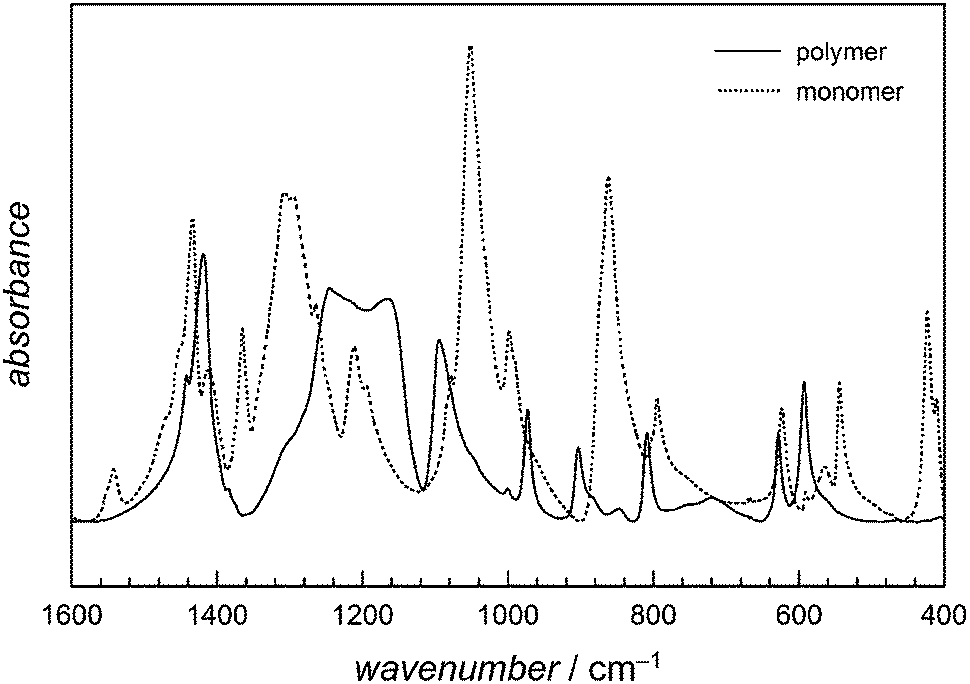 | ||
| Fig. 6 IR spectra of diglycolide and of sample 29 (Mn of 3900 g mol−1). | ||
The thermal properties of the PGA samples were investigated using DSC. The glass transition temperature Tg ranged from 31 to 40 °C and occurred to be independent of Mn. The melting temperature Tm shows a slight variation by 4 °C ranging from 217 to 221 °C. These values are in excellent agreement with values between 220 and 228 °C reported in ref. 33 and 34. As an example, Fig. 7A presents the DSC curve of the PGA sample of Mn = 3900 g mol−1 showing a Tm of (219 ± 2) °C, while both the polymer samples with higher Mn of 13000 and 23100 g mol−1 possess a Tm of (220 ± 2) °C. In all cases the data clearly shows that the thermal properties are almost independent of Mn. Since an increase in Tm with increasing molecular weight is expected for polyesters these results indicate that the critical Mn at which the Tm is a function of Mn should be below 3900 g mol−1.
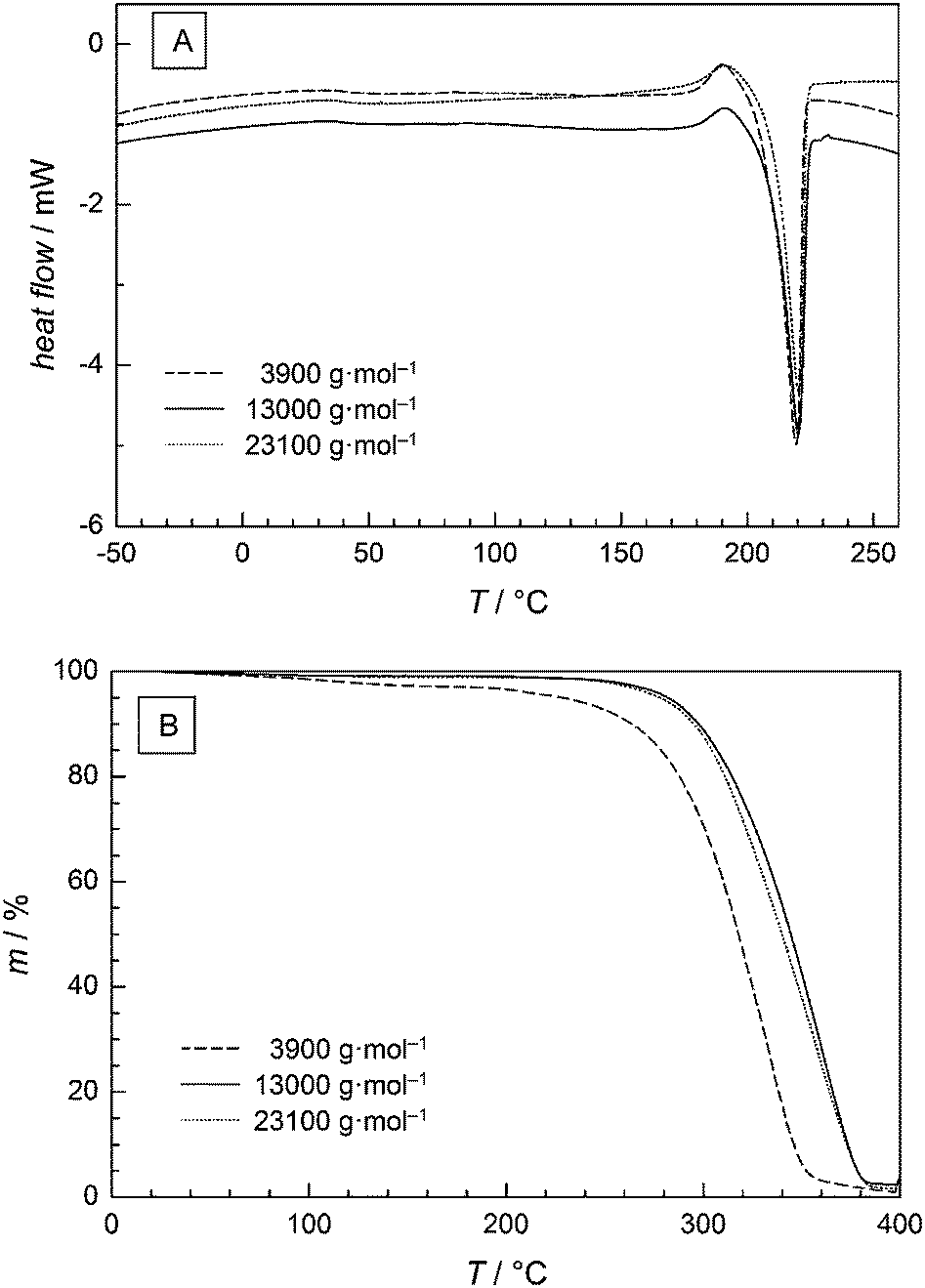 | ||
| Fig. 7 Thermal analyses of PGA samples with different Mn values as indicated: (A) DSC (B) TGA. | ||
The insensitivity of Tm towards Mn can be explained by the high crystallinity detected by WAXS measurements. All samples presented in Fig. 8 show a significant amorphous peak at 2θ = 20° and peaks assigned to the two crystalline phases at 2θ = 23° and 2θ = 29°. Even at low Mn and thereby smaller segments high degrees of crystallinity (DOC) occur. The sample with Mn = 3900 g mol−1 has a DOC of (45.4 ± 0.4)% that is just slightly smaller than for the 13000 g mol−1 sample with DOC of (48.2 ± 0.4)% and the 23100 g mol−1 sample with (46.3 ± 0.7)%. These values are within the range from 40 to 55% reported in literature.35,36
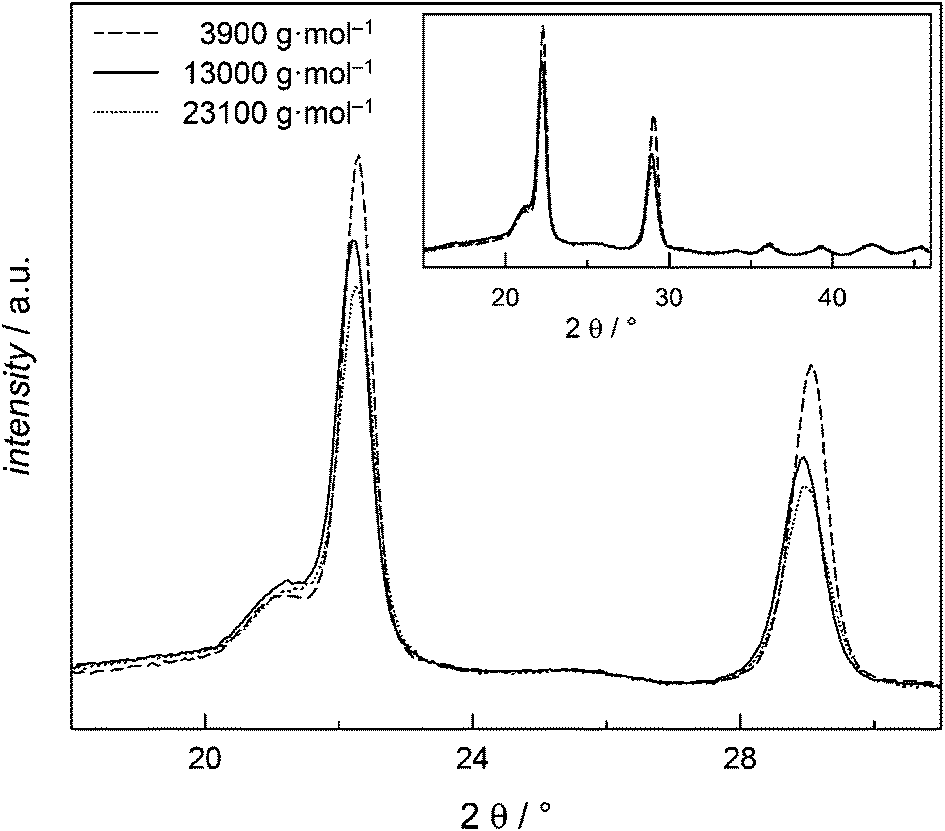 | ||
| Fig. 8 WAXS results for PGA with varying Mn values as indicated. | ||
Furthermore, thermal gravimetric analysis (TGA) was used to study the decomposition behaviour of the materials. Fig. 7B shows that a first decomposition occurs at lower T in case of the low Mn sample. Decomposition occurs most likely by scission of the end groups. The inflection points of the decomposition curve are detected at 310 °C for the low Mn sample and at 340 °C for higher Mn samples. Beyond 360 °C for the low Mn sample and beyond 380 °C for higher Mn samples no further significant weight loss is found. As the decomposition occurs in just one step no intermediate products are generated. Therefore, the data suggests that the decomposition is increased until a certain Mn value is reached. Beyond this Mn no further change in thermal decomposition is found.
With respect to applications the mechanical behaviour of the material is of interest. However, so far the mechanical behaviour could not be investigated. The high-pressure reaction cells yield only small quantities of product, which were not sufficient for the preparation of standardized samples via extrusion and injection moulding. In addition, testing of PGA films was not feasible, because no films suitable for characterization of mechanical properties could be obtained.
4. Conclusions
High molecular weight PGA was synthesized using scCO2 as reaction medium. Within this study the highest Mn of 31200 g mol−1 was obtained at the following reaction conditions: 120 °C and 530 bar with 8.24 × 10−5 mol L−1 catalyst, 6.44 × 10−4 mol L−1 initiator, and 2.88 mol L−1 monomer at a reaction time of 5 hours (sample 12). The data suggests that the molecular weight of the polyester is enhanced by increasing cM. At already comparatively low temperatures and, thus, avoiding thermal decomposition, high molecular weight polymers are accessible. Furthermore, no solvents or any additives, e.g. as surfactants, were involved for the synthesis, which makes this process interesting for degradable polymers intended for biomedical usage.
The use of scCO2 as reaction medium is not only interesting with respect to obtaining a product free of any residues from solvent or additives, it is also attractive for post-polymerization processes. Being able to synthesize PGA in scCO2 constitutes a prerequisite for developing integrated processes combining polymer synthesis, formulation, and shaping of the material.
References
- S. Kaihara, S. Matsumura, P. Fisher and A. Mikos, Nat. Protoc., 2006, 2, 2767–2771 CrossRef PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- J. A. Hyatt, J. Org. Chem., 1984, 49, 5097–5101 CrossRef CAS.
- D. K. Gilding and A. M. Gilding, Polymer, 1979, 20, 1459–1464 CrossRef CAS.
- T. Meyer and M. F. Kemmere, Supercritical Carbon Dioxide in Polymer Reaction Engineering, Wiley-VCH, Weinheim, 2005 Search PubMed.
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207–234 RSC.
- A. I. Cooper, Adv. Mater., 2003, 15, 1049–1059 CrossRef CAS PubMed.
- R. Pini, G. Storti, M. Mazzotti, H. Tai, K. M. Shakesheff and S. M. Howdle, Macromol. Symp., 2007, 259, 197–202 CrossRef CAS PubMed.
- R. Pini, G. Storti, M. Mazzotti, H. Tai, K. M. Shakesheff and S. M. Howdle, J. Polym. Sci., Part B: Polym. Phys., 2007, 46, 483–496 CrossRef PubMed.
- S. Beuermann, M. Buback, M. Gadermann, M. Jürgens and D. P. Saggu, J. Supercrit. Fluids, 2006, 39, 246–252 CrossRef CAS PubMed.
- S. Beuermann, M. Buback and M. Jürgens, Ind. Eng. Chem. Res., 2003, 42, 6338–6342 CrossRef CAS.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–563 Search PubMed.
- H. Tai, W. Wang, R. Martin, J. Liu, E. Lester, P. Licence, H. W. Woods and S. M. Howdle, Macromolecules, 2005, 38, 355–363 CrossRef CAS.
- E. Möller and S. Beuermann, Macromol. React. Eng., 2011, 5, 8–21 CrossRef PubMed.
- A. Galia, G. Caputo, G. Spadaro and G. Filardo, Ind. Eng. Chem. Res., 2002, 41, 5934–5940 Search PubMed.
- I. Idriss, A. Yu, S. M. Howdle, A. K. Whittaker and K. J. Thurecht, Green Chem., 2011, 13, 2032–2037 Search PubMed.
- F. Stassin, O. Halleux and R. J. Jérôme, Macromolecules, 2001, 34, 775–781 CrossRef CAS.
- F. Stassin and R. Jérôme, Chem. Commun., 2003, 232–233 RSC.
- D. Bratton, M. Brown and S. M. Howdle, Macromolecules, 2005, 38, 1190–1195 CrossRef CAS.
- D. D. Hille and M. V. Pishko, Macromol. Rapid Commun., 1999, 20, 511–514 CrossRef.
- R. Mazarro, A. de Lucas, I. García and J. F. Rodríguez, J. Biomed. Mater. Res., Part B, 2008, 85, 196 CrossRef PubMed.
- D. Bratton, M. Brown and S. M. Howdle, Chem. Commun., 2004, 808–809 RSC.
- M. Türk, J. Supercrit. Fluids, 2009, 47, 537–545 CrossRef PubMed.
- E. Reverchon, R. Adami, S. Cardea and G. Della Porta, J. Supercrit. Fluids, 2009, 47, 484–492 CrossRef CAS PubMed.
- M. Buback and C. Hinton, in High-pressure techniques in chemistry and physics: a practical approach, ed. N. Isaacs and W. Holzapfel, Oxford Univ. Press, 1997 Search PubMed.
- M. Tarnacka, T. Flak, M. Dulski, S. Pawlus, K. Adrjanowicz, A. Swinarew, K. Kaminski and M. Paluch, Polymer, 2014, 55, 1984–1990 CrossRef CAS PubMed.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33, 702–709 CrossRef CAS.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef CAS PubMed.
- E. Möller and S. Beuermann, Macromol. React. Eng., 2011, 5, 8–21 CrossRef PubMed.
- C. Kirby and M. A. McHugh, Chem. Rev., 1999, 99, 565–602 CrossRef CAS PubMed.
- C. N. McEwan, C. Jackson and B. S. Larsen, Int. J. Mass Spectrom. Ion Proc., 1996, 160, 387–394 CrossRef.
- C. Jackson, B. Larson and C. N. McEwen, Anal. Chem., 1996, 68, 1303–1308 CrossRef CAS.
- G. Kortaberria, A. Jimeno, P. Arruti, K. de la Caba, P. Remiro, A. Eceiza and I. Mondragon, Macromol. Symp., 2006, 239, 152–158 CrossRef CAS PubMed.
- M. Soccio, N. Lotti, L. Finelli, M. Gazzano and A. Munari, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1901–1910 CrossRef CAS PubMed.
- I. Engelberg and J. Kohn, Biomaterials, 1991, 12, 292–304 CrossRef CAS.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1–16 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06815g |
| This journal is © The Royal Society of Chemistry 2014 |