
Dual effects of mesoscopic fillers on the polyethersulfone modified cyanate ester: enhanced viscoelastic effect and mechanical properties
Zhongnan Hu,
Jie Zhang,
Huiping Wang,
Tian Li,
Zhuoyu Liu and
Yingfeng Yu*
State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai, 200433, China. E-mail: yfyu@fudan.edu.cn; Fax: +86 21 6564 0293; Tel: +86 21 6564 2865
First published on 25th July 2014
Abstract
Phase separation and viscoelastic effect play important roles in the properties of mesoscopic filler reinforced polyethersulfone/cyanate ester composites. In this article, the effects of size and content of mesoscopic fillers on the polymerization induced viscoelastic phase separation of thermoplastic modified thermosets have been studied using optical microscopy (OM), time-resolved light scattering (TRLS) and a rheological instrument. The results of OM and TRLS showed that the characteristic length scale of the phase structure of filler added systems tended to shrink; while the relaxation time of phase separation increased with the decrease of filler size and enlargement of filler content. Rheological behaviours of blends filled with various types and contents of mesoscopic fillers are consistent with the phase separation process. The change of morphology and phase separation process was attributed to the significant enhanced viscoelastic effect of mesoscopic fillers. Both the tensile properties and toughness of cyanate ester blends have been improved due to the “enhanced viscoelastic effect” through addition of mesoscopic fillers. However, the “enhanced viscoelastic effect” of fillers almost disappeared with the diminishing of viscoelastic effect during phase separation.
1. Introduction
Cyanate ester resins have been widely utilized as electronic device encapsulation materials, aerospace structural materials and wave-transparent materials because of their outstanding mechanical, thermal and physical properties.1,2 However, the high brittleness of cyanate ester resins is the main drawback that hinders their application in advanced materials.One of the effective methods to improve the fracture toughness of cyanate esters is to modify them with thermoplastic polymers, such as polysulfone,3,4 poly(ether sulfone),5,6 poly(ether imide)7,8 etc. As the phase structure of blends is vital to fracture toughness,9 it is of great importance to gain control of the phase structure and have a good knowledge of the phase separation process.
The theory of polymerization induced phase separation (PIPS) was first proposed by Inoue et al. in 1980s.10 At the beginning of PIPS, the modifiers (thermoplastic resins) are mixed with thermoset resin monomers to form a homogeneous mixture. During the curing process, the compatibility of the thermoplastic polymer and thermoset resins gets worse due to the growing molecular weight of the thermoset resins, as a result, phase separation takes place and phase structure coarsens with time.
As both the chain mobility and Tg of modifiers and thermosets are quite different from each other, the phase separation process is accompanied with dynamic asymmetry, in which the movement of slow component can't catch up with the phase separation. This kind of “viscoelastic phase separation11–18” was first found by Tanaka et al., and now widely observed in PIPS of thermoplastic–thermoset resin system.19–36
In another aspect, it was found that adding particles into multicomponent system has significant influence on the phase separation process. Tanaka et al.15 have investigated the thermodynamics and kinetics of binary polymer blends with the addition of glass beads, and found that the dispersion of macroscopic fillers is significantly affected by the surface affinity of particles.
Balazs et al.37 considered that nano-fillers would transfer to the interface of the phases due to the enthalpy and the conformation entropy of molecule chains when the scale of the fillers is much smaller than the domain size. Ginzburg38 established a thermodynamic theory model about the phase separation of binary polymer and nano-fillers blends combined with the size of nano-particles and interface effects. Balazs et al.39 predicted that the rate of phase separation and the phase structure were strongly influenced by the addition of nano-fillers according to the multi-field model.
Laradji et al.40,41 found that the development of phase separation is significantly slowed down when the volume fraction and draw ratio of one-dimensional particles were relatively large, because the polymer diffusivity is dramatically decreased due to the kinetic barriers of conformations when the particles gathered in the affinity phase.42
Recently, it was found that adding mesoscopic fillers (meso-fillers), such as carbon nano-tube, nano-metal and whisker, into dynamic asymmetry system, can change the structure and properties of the material substantially.43–48 In our recent work,49,50 it was found that phase structure and phase separation process are significantly affected by viscoelastic effect caused by dynamic asymmetry, and viscoelastic effect can be significantly enhanced due to the addition of inorganic fillers. We called this phenomenon “enhanced viscoelastic effect” in polymerization induced viscoelastic phase separation (PIVPS).
In this article, to make clear the effect of the size and content of meso-fillers in thermoplastics modified thermosets system, and the relationship between phase separation and thermo-mechanical properties, we investigated PIVPS in the polyethersulfone modified cyanate resins system with the addition of one-dimensional meso-fillers in both micron and nano-size (crystal whisker of calcium sulphate and sepiolite), and the phase separation process was prolonged in order to compare with our previous works. In addition, mechanical properties of the blends were also studied with the addition of meso-fillers.
2. Experimental
2.1 Material
The used epoxy oligomer, DER331, was provided by Dow Chemical Co, and is the low molecular weight liquid diglycidyl ether of bisphenol A (DGEBA) with an epoxide equivalent of 182–192. The cyanate used, HF-1, was purchased from Huifeng Co. (Shanghai, China), and is the 4, 4′-cyanate and diphenyl propane (BADCy) with a cyanate equivalent of 139. Polyethersulfone (PES) were supplied by Jilin University, China, has an intrinsic viscosity of 0.43 dL g−1. The crystal whisker of calcium sulphate (CSW), a rod-like mesoscopic inorganic filler with approximately 1 micron in diameter and 10–100 micron in length, was provided by Chengfeng Co., China, without further purification. Sepiolite, a rod-like mesoscopic inorganic filler with approximately 20 nm in diameter and 0.1–2 micrometers in length, was provided by Tolsa (Spain). To ensure good dispersion in polymer blends, the surface of sepiolite was modified by the silane coupling agent 2, 3-epoxy propoxy propyltrimethoxysilicane according to the procedure used in the literature.51 Other chemicals were bought from National drug chemistry Co. China and used without purification.| Sample | PES/BADCy/DGEBA | CSW | Sepiolite | Toluene-p-sulfonic |
|---|---|---|---|---|
| PES-Neat | 12.5/50.2/37.3 | — | — | — |
| PES-Neat-slow | — | — | 0.2 | |
| PES-CSW-2 | 2 | — | — | |
| PES-CSW-2-slow | 2 | — | 0.2 | |
| PES-CSW-4 | 4 | — | — | |
| PES-SEP-2 | — | 2 | — | |
| PES-SEP-2-slow | — | 2 | 0.2 | |
| PES-SEP-4 | — | 4 | — | |
| Neat | 0/50.2/37.3 | — | — | — |
| CSW-2 | 2 | — | — | |
| CSW-4 | 4 | — | — | |
| SEP-2 | — | 2 | — | |
| SEP-4 | — | 4 | — |
Fillers were added to the epoxy oligomer under nitrogen atmosphere with vigorous stirring and ultrasonicating at 100 °C with nominal power output of 360 W for 2 hours and a homogeneous epoxy suspension was obtained. Thermoplastics PES was dissolved in methylene chloride (CH2Cl2) and mixed with epoxy at room temperature. After most of the solvent was evaporated at 60 °C, the epoxy-thermoplastic blends were put under vacuum at 120 °C for 2 h to remove the residual solvent. Then, epoxy mixture and BADCy were blended together with stirring at 90 °C until the mixture was homogeneous. To slower down the curing rate, toluene-p-sulfonic acid was added into the samples with stirring as inhibitor.52 The samples were degassed under vacuum for another few minutes and then cooled to −10 °C to prevent further curing. For mechanical testing, bar samples were cured following the procedure: 140 °C for 1 h, 170 °C for 3 h, and 200 °C for 2 h.
2.2 Experimental techniques
The process of phase separation was tracked by time-resolved light scattering (TRLS) equipped with a controllable hot chamber which was assembled ourselves. The data was recorded every 5 s. The sample blends were pressed into films before TRLS observation.Optical light microscopy (OM) experiments were performed with an Olympus BX51P microscope equipped with an Instec HCS410 hot stage. The sample blends were also pressed into films before OM observation.
The sample blends which were cured isothermally at 450 K were fractured in liquid nitrogen. Then the fracture surface were observed under a scanning electron microscope (SEM, Tescan TS 5163MM). All the samples were coated with gold and mounted on copper mounts.
The melt viscosity variations of the blends during cure reaction were recorded on an Ares-9A rheometry instrument: about 1 g of the blends were sandwiched between two round fixtures and softened at 60 °C for 2 min. The plate distance was then adjusted to about 1 mm, and the temperature was raised quickly at a rate of 100 °C min−1 to the preset curing temperature. All the blends were tested under a parallel plate mode with a controlled strain of 1% and test frequency of 1 Hz.
Tensile tests were measured by Instron 1121 at the constant cross-head speed of 10 mm min−1. Specimens were cured in a dog-bone shape mold. The results were obtained by taking the average values of 5 specimens. The Izod impact test was conducted with a cantilever impact tester XJ-40A at room temperature according to the GB/T 1843-2008.
3. Result and discussion
In this work, we mainly focused on the effect of meso-fillers on the mechanical properties of modified systems. The morphology and meso-fillers effects caused by filler content and particle size are important parameters controlling the structures and mechanical properties of the ternary systems. Influences of these factors were examined.3.1 Mechanical properties and morphology
In this subsection, the stress–strain behaviors and impact strength of meso-fillers added systems with different filler contents and sizes were investigated. Table 2 shows the mechanical properties of the PES modified cyanate blends with meso-fillers. Tensile strength of the blends was improved by the introduction of meso-fillers, and raising meso-fillers amount contributed to the improvement of tensile strength. At the same weight percent of meso-fillers, systems with fillers of smaller size, i.e. sepiolite, had better tensile strength. At the same time, elongation at break of the blends with meso-fillers was also improved.| Samples | PES-Neat | PES-CSW-2 | PES-CSW-4 | PES-SEP-2 | PES-SEP-4 |
|---|---|---|---|---|---|
| Tensile strength (MPa) | 51 ± 4.55 | 57 ± 2.85 | 68 ± 4.40 | 76 ± 3.80 | 81 ± 3.05 |
| Elongation at break (%) | 4.8 ± 0.440 | 5.1 ± 0.255 | 5.8 ± 0.390 | 5.4 ± 0.270 | 5.8 ± 0.290 |
| Impact strength kJ m−2 | 13.7 ± 1.732 | 15.5 ± 0.809 | 16.1 ± 1.366 | 26.5 ± 1.884 | 28.3 ± 2.353 |
As fracture toughness is an important factor that limits the application of cyanate ester resins, the toughness of blends was measured as impact strength in this article. As shown in Table 2, impact strength of PES modified cyanate blends with meso-fillers improved more significantly compared with non-filler systems. It is clear that mechanical properties benefited a lot from the addition of meso-fillers. In addition, the size and content of fillers played an important role in improving the properties of cyanate/PES composites.
As mechanical properties are closely related to their morphology for heterogeneous systems, the phase structure of these blends with and without meso-fillers were compared. Fig. 1 shows the phase structure of these blends after curing. As one can observe from these graphs, phase separation occurred in all the samples but phase structure had great differences between each one. With the increase of filler content and decrease of filler size, the phase structure became finer. Furthermore, it could be observed clearly that meso-fillers tended to immerse into the PES-rich phase. Fig. 1b and c evidently show a well dispersion of CSW in PES-rich phase. From the morphology study, one may expect that the improvement of mechanical properties could be caused by the change of phase structure rather than the general enhanced effect of meso-fillers. To verify this point, the effect caused by phase separation was eliminated by blending cyanate ester resin without PES resin but directly with different meso-fillers. As shown in Table 3, the impact strength declined significantly with the addition of meso-fillers. Obviously, the positive effect caused by meso-fillers in ternary systems shifted into negative effect when there was no interaction between meso-fillers and phase separation.
 | ||
| Fig. 1 SEM graphs of PES modified cyanate blends with the addition of meso-fillers. (a) PES-Neat. (b) PES-CSW-2. (c) PES-CSW-4. (d) PES-SEP-2. (e) PES-SEP-4. | ||
| Samples | Neat | CSW-2 | CSW-4 | SEP-2 | SEP-4 |
|---|---|---|---|---|---|
| Impact strength kJ m−2 | 12.2 ± 1.968 | 6.7 ± 0.634 | 6.6 ± 0.878 | 3.7 ± 0.171 | 2.8 ± 0.856 |
3.2 Effect of filler size on phase separation
As phase structure has great effect on the mechanical properties of cyanate blends with meso-fillers, the difference between CSW and sepiolite added systems should be attributed to the phase separation process, which then dominates the final phase structure of cured materials.Previously, numerous research works have demonstrated that viscoelastic effect had influenced the PIPS in thermoplastic modified thermoset resin system.21,23,24,30–34 In this work, we would further study the PIVPS process of filled and non-filled cyanate ester blends. The phase separation of blend without fillers was first studied to compare the difference between filled systems and unfilled system. Fig. 2a shows a typical evolution process of the PIVPS in the PES/cyanate system (sample PES-Neat) characterized by OM, and similar behaviors have also been observed in numerous literatures.17,19,20,26–30 In Fig. 2a, a micro-bicontinuous phase structure was first observed by OM at the beginning of the phase separation; Along with polymerization the cyanate–epoxy precursors diffused out from the PES-rich phase (dark region) and coarsened rapidly. Then the morphology shifted into a phase inverted structure quickly with small cyanate-rich dispersed particles (bright region) with anisotropic shapes. With the elastic elongation and barking of the PES-rich regions, irregular epoxy-rich macro-phase domains which dispersed in the PES-rich matrix were then formed. Due to the presence of dynamic asymmetry between thermoplastics and thermoset precursors in molecular mobility, molecular weight, and glass transition temperatures, the viscoelasticity of the PES-rich phase as the slower dynamic phase increased with the escape of the thermoset precursor from the thermoplastic-rich phase and eventually behaved as an elastic body.
 | ||
| Fig. 2 Phase separation process of the PES modified cyanate blends without meso-fillers tracked by OM at 395 K. (a) PES-Neat (b) PES-CSW-2 (c) PES-SEP-2. | ||
As the PES chains disentangle much slower than the phase separation, this causes a mechanical stress with great influence on the structure evolution:
| ∂i(Πij − σij + pδij) = 0 | (1) |
To study the influence of filler size on the evolution of phase structure blends filled with one-dimensional meso-fillers, CSW and sepiolite particles were chosen for comparison. The concentrations of particle were first selected as 2 wt% for all the samples. As shown in Fig. 2b, the phase structure of sample PES-CSW-2 was very similar to the sample without any particles (OM results, seen in Fig. 2a) which means the fillers in microscopic had little influence on the phase separation at a relatively low content. When the size of fillers, i.e., sepiolite, was much smaller, cyanate-rich macro-phase domains dispersed in the PES-rich matrix were still formed but with much smaller size (Fig. 2b), which means that the coarsening process of cyanate-rich phase was blocked by the addition of sepiolite. Apparently, the morphology evolution was hindered ominously due to the addition of meso-fillers with smaller size.
It is now well known that dynamic factors rather than thermodynamics factors that govern the distribution of fillers in dynamic asymmetry systems. For sepiolite added systems, we have demonstrated previously that the distribution of sepiolite in slow dynamic phase is caused by “enhanced viscoelastic effect”, which could be well extended to other meso-fillers added systems (as CSW in this article). During the phase separation process, CSW have the lowest mobility and are unable to be transported long distances due to their mesoscale size, and the movement of CSW is also restricted by the entanglement of PES with long molecular chains; although cyanate precursors may have the chance to gather around the CSW due to thermodynamic affinity, the large stress of the PES-rich phase pushes the cyanate diffusing away. The main reason of selective distribution of CSW comes from asymmetric stress division and the large dynamic difference between the immiscible components during phase separation.
From another perspective, the fillers hindered by PES entanglement cause growth in the elastic and viscous moduli of the PES-rich phase resulting in the elongation of the relaxation time of the network. Previous studies have shown that when particles are introduced to high molecular weight polymers, the resulting composites increase their elasticity and viscoelastic moduli, signaling a lengthening of the relaxation time. Fillers distributed in a polymer matrix create confinement of polymer chains and slow down chain dynamics. Entanglements trapped in a confined space become more difficult to relax. As a result, meso-fillers show an “enhanced viscoelastic effect” in all phase separation process with dynamic asymmetry.
To verify the results from OM, TRLS was applied to study the phase evolution process. Fig. 3a shows the TRLS profiles of light scattering vector and intensity with time for samples filled with 2 wt% CSW and sepiolite particles. A relaxation behavior of phase size change was observed by TRLS, as one can see, both of the two filled samples had larger scattering vector qm than that of the sample without particles in Fig. 3a. Furthermore, the light scattering vector of PES-SEP-2 got much larger with phase separation compared to that of PES-CSW-2. In addition, the final qm value increased with the decrease of filler size, which verified the OM observations of smaller characteristic length scale with finer fillers.
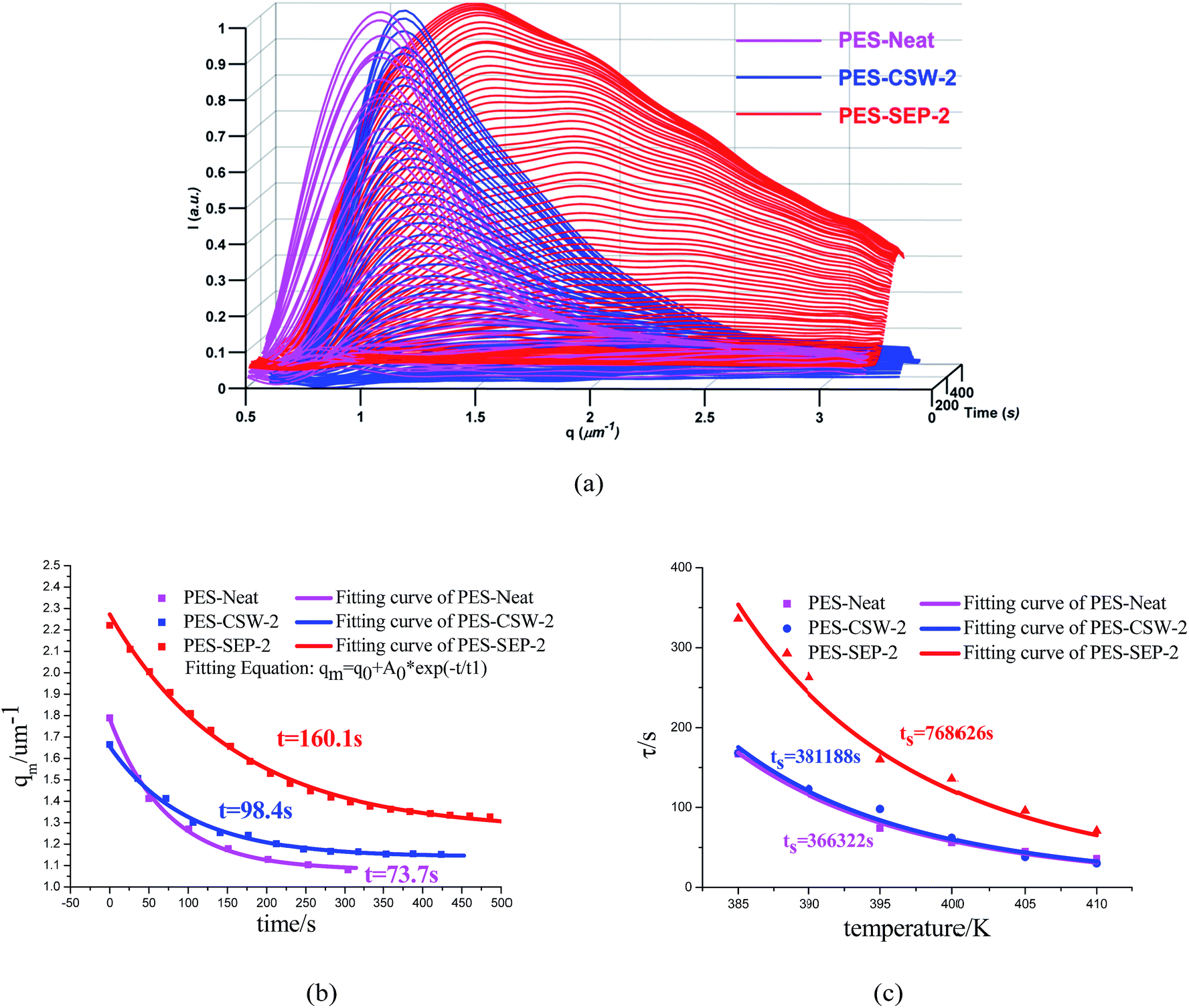 | ||
| Fig. 3 TRLS profile and WLF equation fitting of PES-Neat, PES-CSW-2 and PES-SEP-2 cured at 395 K. (a) Intensity versus q; (b) qm versus time; (c) the relaxation time versus temperature. | ||
Previous and recent works have shown that the evolution of qm corresponds to thermosetting precursor droplets and follows a Maxwell-type relaxation equation (eqn (2)).
| qm(t) = q0 + A1e−t/τ | (2) |
The temperature dependent relaxation time (listed in Table 4) was well fitted by the Williams–Landel–Ferry (WLF) equation according to previous works:21,30,32,33,49,50,53
 | (3) |
| Sample | Relaxation time (s) of different temperatures (K) | |||||
|---|---|---|---|---|---|---|
| 385 | 390 | 395 | 400 | 405 | 415 | |
| PES-Neat | 167 | 121 | 74 | 56 | 45 | 36 |
| PES-CSW-2 | 168 | 123 | 98 | 62 | 38 | 30 |
| PES-CSW-4 | 238 | 163 | 122 | 84 | 56 | 34 |
| PES-SEP-2 | 336 | 263 | 160 | 136 | 96 | 71 |
| PES-SEP-4 | 436 | 307 | 197 | 122 | 81 | 72 |
Taking C1 = 8.86 K and C2 = 101.6 K, the WLF equation can be written as
 | (4) |
As shown in Fig. 3c, all these samples can be fitted quite well by the WLF equation (Fig. 3c). It can be observed that the viscoelastic features of the phase separation do not change with the addition of fillers of different sizes, while the “enhanced viscoelastic effect” gets more notable with the decrease of filler size at the same content.
For meso-fillers of various sizes, one can image that the specific surface area53 increases quickly with the decrease of filler size, which should provide much more entangle points with slow-dynamic phase (PES-chains) during PIVPS, and thus enlarges the dynamics asymmetry. If it is true, i.e., large surface area of meso-fillers favors “enhanced viscoelastic effect”, and increasing the content of fillers (at same level of filler size) would also enlarge the enhancement effect.
 | ||
| Fig. 4 OM morphologies of samples modified by different mass fraction of CSW or sepiolite particles. | ||
For the blends modified by sepiolite particles, with the addition of 4 wt% fillers, the phase structure became much smaller and more refined than the PES/cyanate blend and blend with less fillers. However, the cyanate domains still dispersed in PES matrix with the addition of 4 wt% CSW, the phase size only decreased a little with the increase of filler content for CSW added systems compared to the sepiolite filled sample (Fig. 4).
The TRLS profiles once more showed the same trend with the OM results (seen in Fig. 5). The qm value was strongly affected by the content of sepiolite particles, but changed relatively slightly with CSW.
 | ||
| Fig. 5 TRLS profiles of samples modified by different mass fraction of CSW or sepiolite particles. | ||
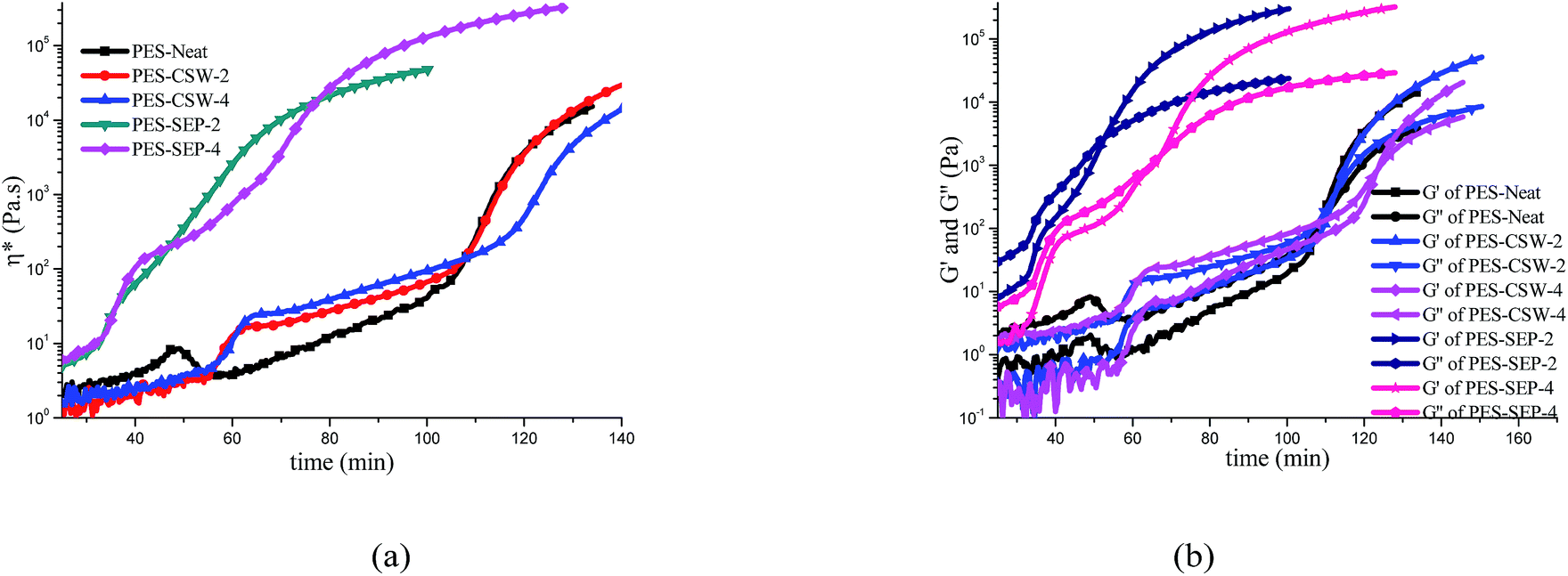 | ||
| Fig. 6 Rheological behavior of the PES modified systems upon curing at 395 K: (a) complex viscosity vs. time; (b) storage and loss modulus vs. time. | ||
Compared to CSW, sepiolite particles have much larger specific surface area; as a result, increasing the content of sepiolite fillers would enlarge the total surface area of fillers greatly, while the surface area of micron-fillers still keeps in a lower level even though their contents are also increased by several times.
The characteristic relaxation time of all the samples calculated from Maxwell-type relaxation equation at all the temperature (listed in Table 4) fitted WLF equation well. Furthermore, the relaxation time at the glass transition temperature obtained from WLF equation increased with the enlarging of surface area (Table 5).
| Sample | PES-Neat | PES-CSW-2 | PES-CSW-4 | PES-SEP-2 | PES-SEP-4 |
|---|---|---|---|---|---|
| R2 | 0.9891 | 0.9567 | 0.9957 | 0.9594 | 0.9837 |
| τs (s) | 366![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 322 322 |
381188 |
519431 |
768626 |
927177 |
Fig. 6a and b show the rheological behavior of all the PES/cyanate systems cured at 395 K; as one can see, the rheological behavior clearly indicates the phase separation and structural transitions. In Fig. 6a, the complex viscosities η* of these blends are plotted at the same curing temperature. Rheological behavior was affected greatly with the addition of sepiolite particles compared with PES-Neat while only changed a little with the addition of CSW, which accorded well with the results of OM and TRLS.
For PES-Neat sample without filler, an increase of viscosity was observed at ca. 45 min (as shown in Fig. 6a), which corresponded to the initial of phase separation. As we know from viscoelastic phase separation systems of thermoplastic-modified thermosets, the phase structures at the initial stage of phase separation are always phase inverted or thermoplastic as the continuous matrix;29,30 therefore, the thermoplastic-rich phase shows an increase in viscosity. However, the PES-Neat system showed a quick drop in viscosity at about 55 min just after the initial increase. This subsequent decrease of viscosity is attributed to the breaking up of thermoplastic matrix structure as the network reverts back to a predominantly cyanate continuous matrix;29,30,54,56,57 while the further growth in viscosity is attributed to the chemical crosslinking of cyanate ester.
The rheological behavior of both PES-CSW-2 and PES-CSW-4 showed similar tendency. At about 55 min (10 min later than PES-Neat) the complex viscosity increased sharply due to the phase separation and formation of bicontinuous structure.30,54–60 The viscosity drop in PES-Neat was not observed because the addition of meso-fillers fixed the phase separation process and kept the bicontinuous structure until the end of evolution. Furthermore, with the increase of surface area, PES-CSW-4 showed larger viscosity growth after phase separation compared to PES-CSW-2. The PES-CSW-4 had a higher viscosity and slightly later gelation time (crossover time of G′ and G′′) compared to PES-Neat and PES-CSW-2.
With the addition of sepiolite particles, complex viscosity sharply increased at about 30 min, and much larger fluctuation happened with the increase of surface area as we expected. The storage and loss modulus corresponded well with the complex viscosity change in each systems, respectively (in Fig. 6b).
Toluene-p-sulfonic acid was added as an inhibitor of cyanate ester resin and thus could lower down the curing rate and prolong the phase separation time, which would effectively reduce the dynamic asymmetry during phase separation.
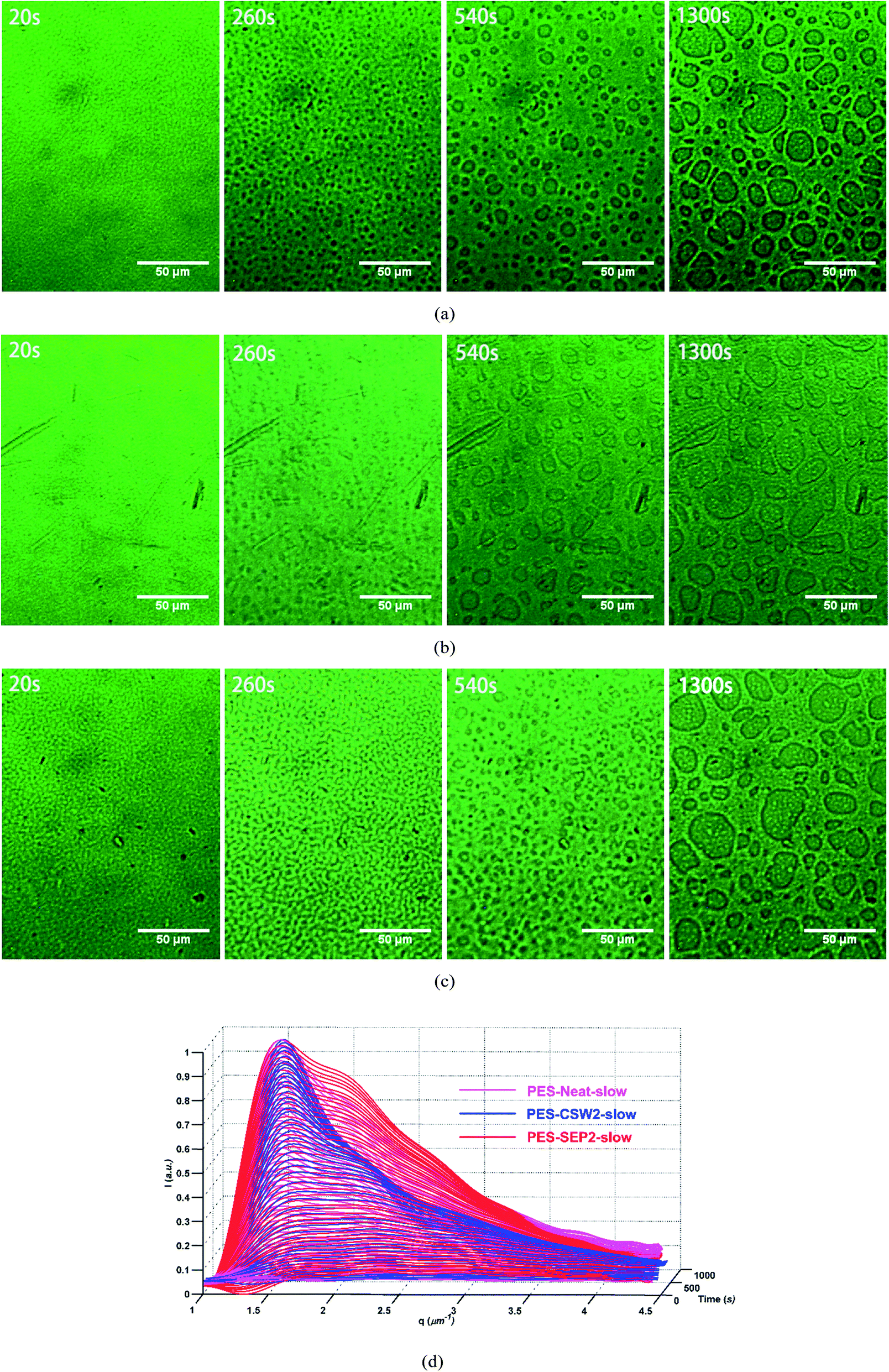 | ||
| Fig. 7 Morphology and TRLS tracking of phase separation process of samples with toluene-p-sulfonic acid cured at 395 K. (a) OM graphs of PES-Neat-slow which was added with toluene-p-sulfonic acid. (b) OM graphs of PES-CSW-2-slow; (c) OM graphs of PES-SEP-2-slow (d) TRLS profiles of samples with toluene-p-sulfonic acid. | ||
Furthermore, the relaxation behavior of PES-CSW-2-slow is nearly the same to that of PES-Neat-slow and PES-SPE-2-slow (Fig. 7d). Obviously, the chain dynamics of PES is no longer slowed down which caused by entanglements with the fillers; and the disentanglement of PES chains is no longer hindered during phase separation. In other words, with low dynamic asymmetry, meso-fillers show limited “enhanced viscoelastic effect” during phase separation.
Conclusions
The mechanical properties of cyanate composites were improved by adjusting phase structure through addition of mesoscopic fillers due to the “enhanced viscoelastic effect” during phase separation. Filler size and content, i.e., surface area of meso-fillers, are important factors that affect the enhanced viscoelastic effect of PIVPS. Larger surface area of fillers provides more entanglement points for slow dynamic chains to interact with meso-fillers and thus significantly enhances the viscoelastic effect. By diminishing dynamic asymmetry, the enhanced viscoelastic effect of meso-fillers tends to disappear due to prolonged chain disentangle time.Acknowledgements
This research work was supported by the National Natural Science Foundation of China (Grant 20974027, 21274031), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.References
- T. Fang and D. A. Shimp, Prog. Polym. Sci., 1995, 20(1), 61–118 CrossRef CAS.
- C. R. Nair, D. Mathew, K. Ninan, New Polymerization Techniques and Synthetic Methodologies, Springer, 2001, pp. 1–99 Search PubMed.
- J. Hwang, K. Cho, T. Yoon and C. Park, J. Appl. Polym. Sci., 2000, 77(4), 921–927 CrossRef CAS.
- I. B. Recalde, D. Recalde, R. García-Lopera and C. M. Gómez, Eur. Polym. J., 2005, 41(11), 2635–2643 CrossRef CAS PubMed.
- J.-Y. Chang and J.-L. Hong, Polymer, 2000, 41(12), 4513–4521 CrossRef CAS.
- J.-Y. Chang and J.-L. Hong, Polymer, 2001, 42(4), 1525–1532 CrossRef CAS.
- I. Harismendy, M. Del Rio, A. Eceiza, J. Gavalda, C. Gomez and I. Mondragon, J. Appl. Polym. Sci., 2000, 76(7), 1037–1047 CrossRef CAS.
- I. Harismendy, M. Del Rio, C. Marieta, J. Gavalda and I. Mondragon, J. Appl. Polym. Sci., 2001, 80(14), 2759–2767 CrossRef CAS PubMed.
- S. Zheng, J. Wang, Q. Guo, J. Wei and J. Li, Polymer, 1996, 37(21), 4667–4673 CrossRef CAS.
- K. Yamanaka, Y. Takagi and T. Inoue, Polymer, 1989, 30(10), 1839–1844 CrossRef CAS.
- Z. He, W. Shi, F. Chen, W. Liu, Y. Liang and C. C. Han, Macromolecules, 2014, 47(5), 1741–1748 CrossRef CAS.
- H. Tanaka, Macromolecules, 1992, 25(23), 6377–6380 CrossRef CAS.
- H. Tanaka and T. Miura, Phys. Rev. Lett., 1993, 71(14), 2244 CrossRef CAS.
- H. Tanaka, J. Chem. Phys., 1994, 100, 5323 CrossRef CAS PubMed.
- H. Tanaka, A. J. Lovinger and D. D. Davis, Phys. Rev. Lett., 1994, 72(16), 2581 CrossRef CAS.
- H. Tanaka, J. Phys.: Condens. Matter, 2000, 12(15), R207–R264 CrossRef CAS.
- H. Tanaka, Faraday Discuss., 2012, 158, 371–406 RSC.
- S. Koizumi, Soft Matter, 2011, 7(8), 3984–3992 RSC.
- Y. Zhang, W. Shi, F. Chen and C. C. Han, Macromolecules, 2011, 44(18), 7465–7472 CrossRef CAS.
- G. Z. Zhan, S. Hu, Y. F. Yu, S. J. Li and X. L. Tang, J. Appl. Polym. Sci., 2009, 113(1), 60–70 CrossRef CAS PubMed.
- W. Gan, Y. Yu, M. Wang, Q. Tao and S. Li, Macromolecules, 2003, 36(20), 7746–7751 CrossRef CAS.
- W. Gan, Y. Yu, M. Wang, Q. Tao and S. Li, Macromol. Rapid Commun., 2003, 24(16), 952–956 CrossRef CAS PubMed.
- S. Goossens, B. Goderis and G. Groeninckx, Macromolecules, 2006, 39(8), 2953–2963 CrossRef CAS.
- J. Jose, K. Joseph, J. r. Pionteck and S. Thomas, J. Phys. Chem. B, 2008, 112(47), 14793–14803 CrossRef CAS PubMed.
- X. Liu, Y. Yu and S. Li, Eur. Polym. J., 2006, 42(4), 835–842 CrossRef CAS PubMed.
- X. Tang, L. Zhang, T. Wang, Y. Yu, W. Gan and S. Li, Macromol. Rapid Commun., 2004, 25(15), 1419–1424 CrossRef CAS PubMed.
- Q. Tao, W. Gan, Y. Yu, M. Wang, X. Tang and S. Li, Polymer, 2004, 45(10), 3505–3510 CrossRef CAS PubMed.
- M. Wang, Y. Yu, X. Wu and S. Li, Polymer, 2004, 45(4), 1253–1259 CrossRef CAS PubMed.
- Y. Yu, M. Wang, D. Foix and S. Li, Ind. Eng. Chem. Res., 2008, 47(23), 9361–9369 CrossRef CAS.
- Y. Yu, M. Wang, W. Gan, Q. Tao and S. Li, J. Phys. Chem. B, 2004, 108(20), 6208–6215 CrossRef CAS PubMed.
- S. Goossens and G. Groeninckx, Macromolecules, 2006, 39(23), 8049–8059 CrossRef CAS.
- P. Jyotishkumar, J. Koetz, B. Tiersch, V. Strehmel, C. Özdilek, P. Moldenaers, R. Hässler and S. Thomas, J. Phys. Chem. B, 2009, 113(16), 5418–5430 CrossRef CAS PubMed.
- P. Jyotishkumar, C. Ozdilek, P. Moldenaers, C. Sinturel, A. Janke, J. r. Pionteck and S. Thomas, J. Phys. Chem. B, 2010, 114(42), 13271–13281 CrossRef CAS PubMed.
- H. Nakanishi, M. Satoh, T. Norisuye and Q. Tran-Cong-Miyata, Macromolecules, 2006, 39(26), 9456–9466 CrossRef CAS.
- P. Jyotishkumar, P. Moldenaers, S. M. George and S. Thomas, Soft Matter, 2012, 8(28), 7452–7462 RSC.
- P. Jyotishkumar, C. Özdilek, P. Moldenaers, C. Sinturel, A. Janke, J. r. Pionteck and S. Thomas, J. Phys. Chem. B, 2010, 114(42), 13271–13281 CrossRef CAS PubMed.
- R. B. Thompson, V. V. Ginzburg, M. W. Matsen and A. C. Balazs, Science, 2001, 292(5526), 2469–2472 CrossRef CAS PubMed.
- V. V. Ginzburg, Macromolecules, 2005, 38(6), 2362–2367 CrossRef CAS.
- A. C. Balazs, V. V. Ginzburg, F. Qiu, G. Peng and D. Jasnow, J. Phys. Chem. B, 2000, 104(15), 3411–3422 CrossRef CAS.
- M. Laradji and M. J. Hore, J. Chem. Phys., 2004, 121, 10641 CrossRef CAS PubMed.
- M. Laradji and G. MacNevin, J. Chem. Phys., 2003, 119, 2275 CrossRef CAS PubMed.
- M. J. Hore and M. Laradji, J. Chem. Phys., 2008, 128, 054901 CrossRef PubMed.
- C. Huang, J. Gao, W. Yu and C. Zhou, Macromolecules, 2012, 45(20), 8420–8429 CrossRef CAS.
- G. Allegra, G. Raos and M. Vacatello, Prog. Polym. Sci., 2008, 33(7), 683–731 CrossRef CAS PubMed.
- D. Gersappe, Phys. Rev. Lett., 2002, 89(5), 058301–058301 CrossRef.
- M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39(16), 5194–5205 CrossRef CAS.
- G. Yu and P. Wu, Polym. Chem., 2013, 5(1), 96–104 RSC.
- W. Shi, X.-M. Xie and C. C. Han, Macromolecules, 2012, 45(20), 8336–8346 CrossRef CAS.
- Y. Liu, X. H. Zhong, G. Z. Zhan, Y. F. Yu and J. Y. Jin, J. Phys. Chem. B, 2012, 116(12), 3671–3682 CrossRef CAS PubMed.
- X. H. Zhong, Y. Liu, H. H. Su, G. Z. Zhan, Y. F. Yu and W. J. Gan, Soft Matter, 2011, 7(7), 3642–3650 RSC.
- P. A. Wheeler, J. Wang, J. Baker and L. J. Mathias, Chem. Mater., 2005, 17(11), 3012–3018 CrossRef CAS.
- I. Hamerton, Chemisty and Technology of Cyanate Ester Resins, Springer Netherlands, 1994 Search PubMed.
- J. Zhang, T. Li, Z. Hu, H. Wang and Y. Yu, RSC Adv., 2014, 4(1), 442–454 RSC.
- A. Bonnet, J. Pascault, H. Sautereau and Y. Camberlin, Macromolecules, 1999, 32(25), 8524–8530 CrossRef CAS.
- A. Bonnet, Y. Camberlin, J. Pascault and H. Sautereau, Macromol. Symp., 2000, 145–150 CrossRef CAS.
- H. Kim and K. Char, Ind. Eng. Chem. Res., 2000, 39(4), 955–959 CrossRef CAS.
- Y. Yu, Z. Zhang, W. Gan, M. Wang and S. Li, Ind. Eng. Chem. Res., 2003, 42(14), 3250–3256 CrossRef CAS.
- A. Tercjak, P. Remiro and I. Mondragon, Polym. Eng. Sci., 2005, 45(3), 303–313 CAS.
- W. Gan, G. Zhan, M. Wang, Y. Yu, Y. Xu and S. Li, Colloid Polym. Sci., 2007, 285(15), 1727–1731 CAS.
- L. Tribut, F. Fenouillot, C. Carrot and J.-P. Pascault, Polymer, 2007, 48(22), 6639–6647 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |