
Spectrofluorimetric and TLC-densitometric methods for a stability indicating assay of valacyclovir hydrochloride in the presence of its degradation product
Sayed M. Derayea,
Islam M. Mostafa and
Mahmoud A. Omar*
Anal. Chem. Dept., Faculty of Pharmacy, Minia University, Minia, Egypt. E-mail: momar71g@yahoo.com
First published on 28th August 2014
Abstract
Two stability-indicating methods were developed and validated for determination of valacyclovir HCl (VAC) in the presence of its degradation product, acyclovir. The first was a TLC-densitometric method, in which chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol:ammonia (50:14:2 v/v/v) was used as a mobile phase. Silica gel 60 F254 was used as a stationary phase and the chromatogram was scanned at 253 nm. Using this chromatographic system, VAC can be readily separated from its degradation product and gives a compact spot at an RF value of (0.55 ± 0.03). The peak area concentration plot is rectilinear over the range 20–300 ng per band. The second method represents the first attempt for spectrofluorimetric determination of VAC. The method was based on the reaction between VAC and 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) in an alkaline medium (pH 8.5) to form a highly fluorescent product that was measured at 500 nm after excitation at 465 nm. The fluorescence intensity concentration plot is rectilinear over the range 1–10 μg ml−1. The proposed methods were successfully applied for the determination of VAC in its commercial tablets with average percentage recovery of 100.13 ± 0.33 and 98.50 ± 1.75 for TLC-densitometric and spectrofluorimetric methods, respectively, without interference from common excipients. The results of the proposed methods were statistically analyzed and found to be in accordance with those given by a reported method. In addition the proposed methods were extended to a stability study of VAC, where the drug was exposed to acidic, alkaline, oxidative and photolytic degradation according to ICH guidelines.
:methanol:ammonia (50:14:2 v/v/v) was used as a mobile phase. Silica gel 60 F254 was used as a stationary phase and the chromatogram was scanned at 253 nm. Using this chromatographic system, VAC can be readily separated from its degradation product and gives a compact spot at an RF value of (0.55 ± 0.03). The peak area concentration plot is rectilinear over the range 20–300 ng per band. The second method represents the first attempt for spectrofluorimetric determination of VAC. The method was based on the reaction between VAC and 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) in an alkaline medium (pH 8.5) to form a highly fluorescent product that was measured at 500 nm after excitation at 465 nm. The fluorescence intensity concentration plot is rectilinear over the range 1–10 μg ml−1. The proposed methods were successfully applied for the determination of VAC in its commercial tablets with average percentage recovery of 100.13 ± 0.33 and 98.50 ± 1.75 for TLC-densitometric and spectrofluorimetric methods, respectively, without interference from common excipients. The results of the proposed methods were statistically analyzed and found to be in accordance with those given by a reported method. In addition the proposed methods were extended to a stability study of VAC, where the drug was exposed to acidic, alkaline, oxidative and photolytic degradation according to ICH guidelines.
1. Introduction
Valacyclovir hydrochloride (VAC) is L-valine 2-[(2-amino-1,6-dihydro-6-oxo-9H-purin-9yl)methoxy]ethyl ester hydrochloride (Fig. 1). After its oral administration, VAC is rapidly converted into acyclovir which has demonstrated antiviral activity, against herpes simplex virus type I (HSV-1), type 2 (HSV-II) and Varicella zoster virus.1 The oral bioavailability of acyclovir is higher after administration of VAC relative to acyclovir itself.2,3 The mechanism of action of acyclovir involves the highly selective inhibition of virus DNA replication, via enhanced uptake in virus-infected cells and phosphorylation by viral thymidine kinase.4,5 Few analytical methods have been reported for the determination of VAC. These methods include: spectrophotometric,6–8 chromatographic,9–22 capillary electrophoretic23 and electrochemical methods.24 To the present time there is no spectrofluorometric method reported for analysis of VAC. Accordingly we here present the first attempt for spectrofluorimetric method in addition to TLC-densitometry for the determination of VAC in pure form and pharmaceutical tablets. TLC-densitometric method is becoming a routine technique for analysis of drugs due to its low operating cost, high sample-throughput, and need for minimum sample clean-up. The major advantage of TLC-densitometric lies in several samples can be run simultaneously using a small quantity of mobile phase thus lowering analysis time and cost per analysis, Unlike HPLC, wherein substantial amounts of mobile phase and time are required for analysis of multiple samples.25 TLC-densitometric method has the advantages of being sensitive, cheap and one of the simplest chromatographic technique that does not need large precautions as in HPLC technique which should be taken to prevent several problems in HPLC such as plugging of the column and variation of retention time. Both proposed methods of analysis of the cited drug are able to determine the drug in pure form and pharmaceutical tablets without interference from tablet excipients. Furthermore, both methods were developed to be stability-indicating methods that can be used for determination of VAC in presence of its degradation products.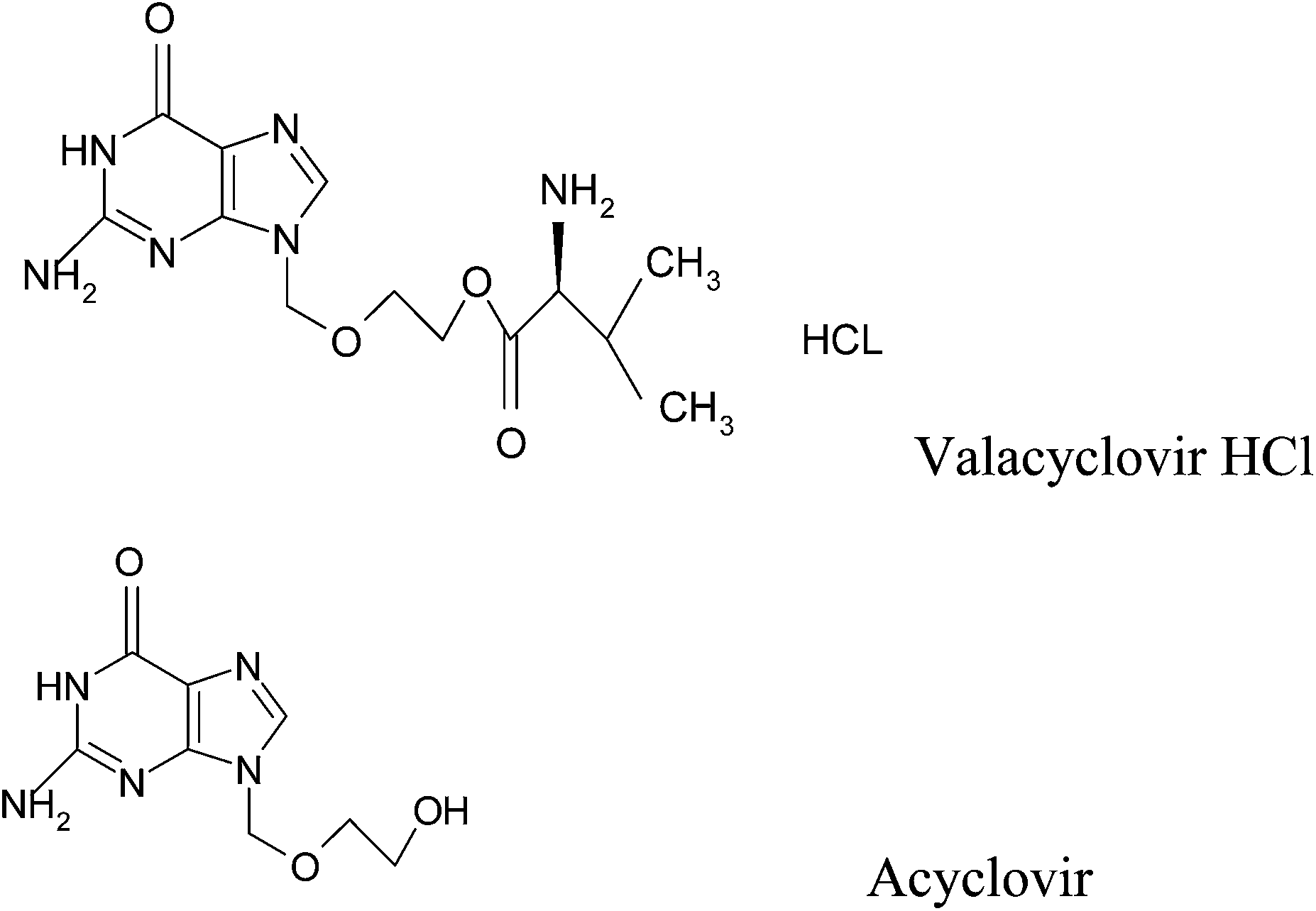 | ||
| Fig. 1 Chemical structure of valacyclovir HCl and acyclovir. | ||
2. Experimental
2.1. Apparatus
Camag TLC scanner 3 S/N 130319 in the absorbance mode utilizing deuterium lamp as a source of radiation with Wincats software. Samples were applied with Camag micro-syringe 100 μl, using a Camag Linomat 5 autosampler, (Camag, Muttenz, Switzerland).A Perkin Elmer LS 45 luminescence spectrometer (United Kingdom) equipped with 150 watt xenon arc lamp and 1 cm quartz cell. Slit width for both monochromators were set at 10 nm. The spectrometer is connected to a PC loaded with the FL Winlab™ software, Spectronic™ Genesys™ 2 PC. Ultraviolet/visible spectrophotometer (Milton Roy Co, USA) with matched 1 cm quartz cell and was connected to PC loaded with Winspec™ application software.
MLW type thermostatically controlled water bath (Memmert GmbH, Schwabach, Germany) was used for heating purposes.
2.2. Material and chemicals
VAC was obtained as a gift from (Hikma Pharma, Cairo, Egypt). Valtovir® tablets (Hikma Pharma, Cairo, EGYPT) labeled to contain 1110 mg VAC per tablet was purchased from local market. Acyclovir was obtained as a gift from GlaxoSmithKline (Cairo, Egypt). 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole NBD-Cl (Sigma Chemical Co., St. Louis, USA) was freshly prepared as 0.1% w/v in methanol. Chloroform, hydrochloric acid, ammonia solution, hydrogen peroxide (30% v/v), sodium hydroxide, boric acid and methanol were purchased from El-Nasr Chemical Co. (Cairo, Egypt). All chemicals and reagents used were of analytical grade.Aluminum plate precoated with silica gel 60 F254, (20 × 10 cm) with 250 μm thickness (Merck, Darmstadt, Germany).
2.3. Standard solution preparation
An accurately weighed amount (10 mg) of VAC was quantitatively transferred into a 100 ml calibrated flask, dissolved in 20 ml methanol, and then completed to volume with the same solvent to obtain a stock solution of 0.1 mg ml−1. The stock solution was further diluted with methanol to obtain working solutions in the concentration range of 5–75 μg ml−1 and 10–100 μg ml−1 for TLC-densitometric and spectrofluorimetric methods, respectively.2.4. Preparation of sample solutions
An accurately weighed amount of finely powdered twenty tablets equivalent to 25 mg of VAC was transferred into a 50 ml volumetric flask. The powder was dissolved in 25 ml methanol, sonicated for 15 min, diluted to the mark with methanol and mixed well. The resulting solution was centrifuged at 3000 rpm for 5 min. Two ml of this solution was diluted to 10 ml with methanol.2.5. General analytical procedure
:methanol:ammonia (50:14:2 v/v/v) as a mobile phase was performed in a Camag twin-trough glass chamber previously saturated with mobile phase for 15 min at room temperature. After development, the plates were dried in a current of hot air using a hair-dryer. Densitometric scanning in reflectance mode at 253 nm was performed at 20 mm s−1 scanning speed and the slit dimension was kept at 6.0 × 0.30 μm. The TLC chromatograms were manipulated by WINCATS software.2.6. Procedures for stability indicating assay
Oxidative induced degradation with hydrogen peroxide was carried out by transferring 1.0 ml of VAC solution working solution (1 mg ml−1) into 10 ml volumetric flask then 1 ml of hydrogen peroxide (30.0% v/v) was added. The solution was kept at room temperature for a period of 2 hours. The solution was heated in boiling water bath for 10 minutes to completely remove the excess of hydrogen peroxide. The volume was completed to 10 ml with methanol.
The photochemical stability of the drug was studied by exposing the stock solution (0.1 mg ml−1) to direct artificial day light for five days.
For TLC-densitometric method 6 ml of the above solution was diluted to 10 ml with methanol in volumetric flask then 4 μl was applied to TLC plate and the plate was developed and analyzed as described in “general analytical procedures”. For spectrofluorimetric, 1.0 ml of the above solution was used in the “general analytical procedures”.
3. Results and discussion
3.1. Method optimization for the TLC-densitometric measurements
TLC procedure was optimized with a view to develop a stability-indicating assay method. The pure forms of drug and its degradation product, acyclovir were spotted on TLC plates and run in different solvent systems. Initially, chloroform–methanol in varying ratios was tried. Chloroform–methanol (5:1.4 v/v) as a mobile phase gave good resolution with RF value of 0.56 ± 0.03 for VAC and 0.3 ± 0.03 for acyclovir unfortunately with tailing peaks. Addition of 0.2 ml ammonia to the above mobile phase gave compact spots for both drug and acyclovir. The final mobile phase consisted of chloroform:methanol:ammonia (5:1.4:0.2 v/v/v) which gave a sharp and symmetrical peaks. To obtain a well-defined spots the chamber was saturated with the mobile phase for 15 min at room temperature prior to the analysis.
3.2. Method optimization for the spectrofluorimetric measurements
VAC contains a primary amino group that could react with NBD-Cl through a condensation reaction. This reaction was investigated in this study to develop the first spectrofluorimetric method for determination of VAC. The reaction product was colored yellow and exhibited highest fluorescence intensity at λem 500 nm after excitation at λex 465 nm, (Fig. 2). The experimental parameters affecting the development and stability of the reaction product were investigated and optimized. Each parameter was changed individually while the others were kept constant. These parameters include; pH, volume of the buffer, concentration of NBD-Cl, reaction time, temperature and diluting solvent. | ||
| Fig. 2 Excitation and emission spectra of the reaction between NBD-Cl and VAC (4 μg ml−1). | ||
 | ||
| Fig. 3 Effect of the pH on the condensation reaction of VAC (4 μg ml−1) and NBD-Cl. | ||
 | ||
| Fig. 4 Effect of the volume of NBD-Cl reagent on the fluorescence intensity of the reaction with VAC (4 μg ml−1). | ||
3.3. Stoichiometry and mechanism of the reaction
Job's method of continuous variation28 was employed for investigating the stoichiometry of the reaction. Equimolar solutions of both the drug and NBD-Cl reagent (1 × 10−3 M) were prepared. Then portions of mixture of master solutions of the drug and reagent were made up comprising different complementary proportions (0.2:2.8, 0.4:2.6, 0.5:2.5…2.6:0.4, 2.8:0.2). Results suggested that VAC reacts with NBD-Cl in the ratio 1:1 (Fig. 5). Based on this ratio the suggested mechanism of the condensation reaction with NBD-Cl was presented in (Fig. 6).
 | ||
| Fig. 5 The stoichiometry of the reaction between NBD-Cl and VAC. | ||
 | ||
| Fig. 6 The suggested mechanism of the condensation reaction of VAC with NBD-Cl. | ||
It should be noted that, the degradation product, acyclovir could not undergo the condensation reaction with NBD-Cl and this can be inferred from the absence of any fluorescence property due to the reaction. This may be attributed to tautomerism of the amino group in the guanine base making the proton of amino group acidic therefore no condensation reaction could occur with NBD-Cl.
3.4. Method validation
The proposed methods have been validated according to ICH guidelines.29 The investigated parameters include: linearity and range, sensitivity, precision, accuracy, recovery and robustness.The International Conference on Harmonization (ICH) guideline entitled ‘stability testing of new drug substances and products’ requires the stress testing to be carried out to elucidate the inherent stability characteristics of the active substance.30 An ideal stability-indicating method is one that quantifies the drug and resolves its degradation products.
| Parameters | TLC-densitometric method | Spectrofluorimetric method |
|---|---|---|
| a Number of determination is 9 for TLC-densitometric method and 7 for spectrofluorimetric method.b At 95% confidence limit. | ||
| Linearity range | 20–300 (ng per band) | 1–10 (μg ml−1) |
| Correlation coefficient (r) | 0.9996 | 0.9996 |
| Determination coefficient (r2) | 0.9992 | 0.9991 |
| Intercepta ± SD | 249.75 ± 24.31 | 104.03 ± 1.67 |
| Confidence limit of interceptb | 192.25 | 99.74 |
| Slopeb ± SD | 12.48 ± 0.14 | 22.2 ± 0.29 |
| Confidence limit of slopeb | 12.16 | 21.45 |
| LOD | 6.42 (ng per band) | 0.25 (μg ml−1) |
| LOQ | 19.47 (ng per band) | 0.75 (μg ml−1) |
| TLC-densitometric method | Spectrofluorimetric method | ||||
|---|---|---|---|---|---|
| Concentration (ng per band) | % Recoverya ± SD | Concentration (μg ml−1) | % Recoverya ± SD | ||
| Taken | % Added | Taken | % Added | ||
| a The value is the average of six determinations. | |||||
| 80 | 0 | 99.56 ± 0.46 | 2 | 0 | 99.74 ± 1.29 |
| 80 | 50% | 99.70 ± 0.21 | 2 | 50% | 99.10 ± 0.99 |
| 80 | 100% | 99.85 ± 0.11 | 2 | 100% | 98.55 ± 0.69 |
| 80 | 150% | 99.90 ± 0.12 | 2 | 150% | 99.35 ± 0.45 |
| Intra-day precision | Inter-day precision | ||
|---|---|---|---|
| Amounta | % Recoveryb ± % RSD | Amounta | % Recoveryb ± % RSD |
| a The amount is ng per band for TLC-method and μg ml−1 for the spectroflourimetric method.b The value is the average of six determinations. | |||
| TLC-densitometric method | |||
| 80 | 99.89 ± 0.33 | 0.08 | 98.41 ± 0.29 |
| 120 | 98.60 ± 0.52 | 0.12 | 99.73 ± 0.26 |
| 300 | 99.45 ± 0.49 | 0.30 | 99.55 ± 0.39 |
| Spectrofluorimetric method | |||
| 3 | 99.07 ± 1.28 | 3 | 99.86 ± 1.22 |
| 4 | 101.2 ± 0.26 | 4 | 99.32 ± 0.45 |
| 6 | 100.8 ± 1.11 | 6 | 98.65 ± 0.93 |
Robustness of TLC-densitometric method was performed by introducing small changes in the mobile phase composition, saturation time and wavelength, then examining their effects on the obtained results. Mobile phases having different composition of chloroform:methanol:ammonia (4.9:1.3:0.1 and 5.1:1.5:0.3, v/v/v) and different wavelengths (248 and 258 nm). Robustness for spectrofluorimetric method was performed by making small changes in general analytical procedure, such as NBD-Cl volume (±0.2 ml), solution pH (±0.2 unit), and volume of the buffer (±0.5 ml). The obtained results from small variation in any of these parameters did not significantly affect the results of both methods. Therefore the procedures of the proposed methods are considered robust.
3.5. Stability-indicating studies
VAC was exposed to different stress conditions including acid, alkali, oxidation and photo-degradations. After applying the degradation procedures, the proposed methods were applied for determination of the extent of degradation of the cited drug. Alkaline and Acidic induced hydrolysis resulted in the appearance of an additional peak at RF value of 0.3 ± 0.03 (Fig. 7a and b). The new peak appears before the parent drug peak which gives an indication that the degradation product is more hydrophilic and contains polar group. The degradant was identified as acyclovir through comparison between RF of the degradant and RF of the standard authentic acyclovir. About 45% of the drug was degraded in case of acidic degradation while 52% of the drug was degraded in case of alkaline degradation. On the other hand, the exposure to oxidation with 30% v/v hydrogen peroxide and photodegradation with direct sun light for five days did not produce any significant effect on the drug chromatograms and only the peak of VAC was observed at RF = 0.55 (Fig. 7c and d). This gives an indication about the stability of VAC against oxidation and photodegradation but its liability of for basic and acidic hydrolysis. | ||
| Fig. 7 TLC chromatograms of VAC degradation. (a) Alkali induced degradation. (b) Acid induced degradation. (c) Photolytic degradation. (d) Oxidative degradation. | ||
3.6. Application to tablets
The proposed methods were applied for determination of VAC in the commercially available tablets. The mean recoveries values were 100.13 ± 0.33 and 98.5 ± 1.75 for TLC-densitometric and spectrofluorimetric methods, respectively. The results of the proposed methods were statistically compared with that of the reference method8 regarding t- and F- tests at 95% confidence level. As shown in Table 4, there is no significant difference between the results of both proposed and reported method, as the calculated values is less than the tabulated one. This indicated that the proposed methods have good accuracy and precision.4. Conclusion
The proposed TLC-densitometric and spectrofluorimetric methods are accurate, sensitive and selective methods. They allow the determination of VAC in pure form and in the presence of its degradation product, as well as in pharmaceutical dosage form. So, it can be considered as a stability-indicating one. The satisfactory sensitivity and simplicity make the methods suitable for routine analysis of VAC in quality control laboratories.References
- Martindale: the Complete Drug Reference, S. C. Sweetman (ed.), Pharmaceutical Press, London, 33rd edn, 1999, p. 643 Search PubMed.
- D. D. Phan, P. Chin-Hong, P. E. T. Lin, P. Anderle, W. Sadee and B. J. Guglielmo, Intra- and interindividual variabilities of valacyclovir oral bioavailability and effect of coadministration of an hPEPT1 inhibitor, Antimicrob. Agents Chemother., 2003, 47, 2351–2353 CrossRef CAS.
- A. E. Thomsen, M. S. Christensen, M. A. Bagger and B. Steffansen, Acyclovir prodrug for the intestinal di/tri-peptide transporter PEPT1: comparison of in vivo bioavailability in rats and transport in Caco-2 cells, Eur. J. Pharm. Sci., 2004, 23, 319–325 CrossRef CAS PubMed.
- J. J. O'Brien and D. M. Campoli-Richards, Acyclovir. an updated review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy, Drugs, 1989, 37, 233–309 CrossRef PubMed.
- C. P. Landowski, D. Sun, D. R. Foster, S. S. Menon, J. L. Barnett, L. S. Welage, C. Ramachandran and G. L. Amidon, Gene expression in the human intestine and correlation with oral valacyclovir pharmacokinetic parameters, J. Pharmacol. Exp. Ther., 2003, 306, 778–786 CrossRef CAS PubMed.
- D. V. P. Reddy, B. M. Gurupadayya and Y. N. Manohara, Spectrophotometric determination of valacyclovir hydrochloride in bulk and pharmaceutical formulations, Asian J. Chem., 2007, 19, 2797–2800 CAS.
- G. S. Babu, I. S. Babu and N. K. Kumar, Spectrophotometric determination of valacyclovir in pharmaceutical formulation, Asian J. Chem., 2007, 19, 1642–1644 CAS.
- M. Ganesh, C. V. Narasimharao and A. S. Kumar, UV spectrophotometric method for the estimation of valacyclovir hcl in tablet dosage form, E–J. Chem., 2009, 6, 814–818 CrossRef CAS PubMed.
- M. L. Palacios, G. Demasi and A. T. Pizzorno, Validation of an HPLC method for the determination of valacyclovir in pharmaceutical dosage, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 751–762 CrossRef CAS PubMed.
- P. N. Rao, K. R. Rajeswari and V. J. Rao, RP-HPLC estimation of valacyclovir in tablets, Asian J. Chem., 2006, 18, 2552–2556 CAS.
- R. Kanneti, R. Rajesh, J. A. Raj and P. A. Bhatt, An LC–MS–MS Method for the Simultaneous Quantitation of Acyclovir and Valacyclovir in Human Plasma, Chromatographia, 2009, 70, 407–414 CAS.
- J. J. Sasanya, A. M. M. Abd-Alla and A. G. Parker, Analysis of the antiviral drugs acyclovir and valacyclovir-hydrochloride in tsetse flies (Glossina pallidipes) using LC-MSMS, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2384–2390 CrossRef CAS PubMed.
- C. Pham-Huy, F. Stathoulopoulou and P. Sandouk, Rapid determination of valaciclovir and acyclovir in human biological fluids by high-performance liquid chromatography using isocratic elution, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 732, 47–53 CrossRef CAS.
- A. Savaser, C. K. Ozkan and Y. I. Ozkan, Development and validation of an RP-HPLC method for the determination of valacyclovir in tablets and human serum and its application to drug dissolution studies, J. Liq. Chromatogr. Relat. Technol., 2003, 26, 1755–1767 CrossRef CAS PubMed.
- A. S. Jadhav, D. B. Pathare and M. S. Shingare, Development and validation of enantioselective high performance liquid chromatographic method for valacyclovir, an antiviral drug in drug substance, J. Pharm. Biomed. Anal., 2007, 43, 1568–1572 CrossRef CAS PubMed.
- K. Maria, G. Evagelos, G. Sofia, K. Michael and P. Irene, Selective and rapid liquid chromatography/negative-ion electrospray ionization mass spectrometry method for the quantification of valacyclovir and its metabolite in human plasma, J. Chromatogr. B., 2008, 864, 78–86 CrossRef PubMed.
- Y. Manish, U. Vivek, S. Puran, G. Sailendra and S. S. Pranav, Stability evaluation and sensitive determination of antiviral drug, valacyclovir and its metabolite acyclovir in human plasma by a rapid liquid chromatography-tandem mass spectrometry method, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 680–688 CrossRef PubMed.
- P. V. M. K. Gowtham, C. Jothimanivannan, C. H. Arjun, M. Jambulingam, S. Ananda Thangadurai and D. Kamalakannan, Accelerated Stability Studies on Valacyclovir Hydrochloride by RP-HPLC, Int. J. Pharm., Chem. Biol. Sci., 2013, 2, 110–117 Search PubMed.
- G. S. Holkar, M. D. Rokade, R. R. Yadav and V. N. Daphal, Determination of Assay and Peak Purity Evaluation of Acyclovir and Valacyclovir by RP-HPLC Method, Int. J. Theor. Appl. Sci., 2012, 4, 56–65 CAS.
- S. Yasmeen, K. A. Nanda and K. P. Safia, Development and Validation of Stability Indicating RP-HPLC Method for Estimation of Valacyclovir in Pharmaceutical Dosage Forms, Int. J. Phar. Clin. Res., 2013, 5, 7–12 Search PubMed.
- B. Jahnavi and G. Ashok, Method Development and Validation of Valacyclovir Hydrochloride Assay by Rp-Hplc, in Pharmaceutical Dosage Form, 2009, vol. 1, pp. 16–26 Search PubMed.
- K. R. Srinivasa and M. Sunil, Stability-indicating liquid chromatographic method for valacyclovir, Int. J. ChemTech Res., 2009, 1, 702–708 Search PubMed.
- K. M. Al Azzam, B. Saad, A. Makahleah, H. Y. Aboul-Enein and A. A. Elbashir, Assay and stability-indicating micellar electrokinetic chromatography method for the simultaneous determination of valacyclovir, acyclovir and their major impurity guanine in pharmaceutical formulations, Biomed. Chromatogr., 2010, 24, 535–543 CAS.
- B. Uslu, S. A. Özkan and Z. Şentürk, Electrooxidation of the antiviral drug valacyclovir and its square-wave and differential pulse voltammetric determination in pharmaceuticals and human biological fluids, Anal. Chim. Acta, 2006, 555, 341–347 CrossRef CAS PubMed.
- M. P. Shubhangi and R. D. Sunil, Validated High-Performance Thin-Layer Chromatographic Method for Quantitation of Fluvoxamine in the Presence of Degradation Products Formed under ICH Recommended Stress Conditions, J. Planar Chromatogr.--Mod. TLC, 2012, 25, 338–343 CrossRef.
- H. Miyano, T. Toyo'oka and K. Imai, Further studies on the reaction of amines and proteins with 4-fluoro-7-nitrobenzo-2-oxa-1,3-diazole, Anal. Chim. Acta, 1985, 170, 81–87 CrossRef CAS.
- K. Imai, T. Toyo'oka and H. Miyano, Fluorigenic reagents for primary and secondary amines and thiols in high performance liquid chromatography: a review, Analyst, 1984, 109, 1365–1373 RSC.
- P. Job, Advanced Physicochemical Experiments, Oliner and Boyed, Edinburgh, 2nd edn, 1964, p. 54 Search PubMed; P. Job, Ann. Chem., 1936, 16, 97 Search PubMed.
- ICH Harmonized Tripartite Guideline Q2 (R1), Validation of analytical procedures: text and methodology, Current step 4 version, 2005.
- ICH, Q2B Validation of Analytical Procedure: Methodology International Conference on Harmonization, Geneva, March 1996 Search PubMed.
- The United States Pharmacopoeia XXV and NF XX, American Pharmaceutical Associtation, Washington, DC, 2002 Search PubMed.
- ICH Harmonized Tripartite Guideline, Topic Q2A: Text on validation of analytical procedure, 1994.
| This journal is © The Royal Society of Chemistry 2014 |