
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-based synthesis and cleaning methods for high purity ZnO nanoparticles – comparing acetate, chloride, sulphate and nitrate zinc salt precursors†
A. M. Pourrahimia,
D. Liua,
L. K. H. Pallona,
R. L. Anderssona,
A. Martínez Abadb,
J.-M. Lagarónb,
M. S. Hedenqvista,
V. Strömc,
U. W. Geddea and
R. T. Olsson*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Fibre and Polymer Technology, SE-100 44 Stockholm, Sweden. E-mail: rols@kth.se; Fax: +46 8 208856; Tel: +46 8 7907637
bInstitute of Agrochemistry and Food Technology (IATA), Novel Materials and Nanotechnology Group Valencia, Spain
cKTH Royal Institute of Technology, School of Industrial Engineering and Management, Material Science and Engineering, SE-100 44 Stockholm, Sweden
First published on 1st August 2014
Abstract
A low temperature (60 °C) aqueous synthesis method of high purity ZnO nanoparticles intended as fillers for ultra-low electrical conductivity insulations is described. Particles were prepared under identical conditions from different zinc salts based on nitrate, chloride, sulphate or acetate to compare their abilities to form high yields of sub-50 nm particles with narrow size distribution. The acetate salt gave uniform 25 nm ZnO particles with a conical prism shape. The chloride and sulphate derived particles showed mixed morphologies of nanoprisms and submicron petals, whereas the nitrate salt yielded prisms assembled into well-defined flower shapes with spiky edges. The micron-sized flower shapes were confirmed by X-ray diffraction to consist of the smaller prism units. Photoluminescence spectroscopy showed emission in the blue-violet region with little variation depending on precursor salt, suggesting that the spectra were dependent on the primary nanoprism formation and rather independent of the final particle morphology. Microscopy revealed that the salt residuals after the reaction showed different affinity to the particle surfaces depending on the type of salt used, with the acetate creating ca. 20 nm thick hydrated shells; and in falling order of affinity: chloride, sulphate and nitrate. An acetate ion shielding effect during the synthesis was therefore assumed, preventing nanoparticle fusion during growth. Varying the concentrations of the counter-ions confirmed the shielding and only the acetate anions showed an ability to stabilize solitary nanoprisms formation in reaction yields from 2 to 10 g L−1. Ultrasonic particle surface cleaning was significantly more efficient than water replacement, resulting in a stable aqueous dispersion with a high zeta potential of 38.9 mV at pH 8.
1. Introduction
Wurtzite crystals of zinc oxide (ZnO) have a large band gap (3.37 eV) with an excitation energy of 60 meV. Together with its piezoelectric properties, this has made ZnO an attractive material for existing and emerging applications such as voltage arrestors (varistors), acoustic devices, UV radiation devices, magneto-optical devices, chemical and biological sensing, energy storage and conversion (photovoltaic cells, batteries, capacitors and hydrogen storage devices), separation catalysts, hybrid LEDs, electrochromic displays and high-voltage insulations.1–8,10 In many of these applications, ZnO nanoparticles are of interest due to their increasing surface-to-volume ratio with decreasing particle size, combined with the possibility of oriented crystal growth yielding anisotropic particles.3,4,7 In particular, nano-sized particles are interesting in various polymer composite formulations for transparent electronics and UV-absorption applications,4 with the advantage that narrowly nanosized particles in a polymer matrix improve the mechanical properties of the composite. Tensile strength of nitrile butadiene rubber and natural rubber was increased ca. 70 and 80%, respectively, when nanoparticles were used instead of μm-sized particles.9 Recently, ultra-low electrical conductivity nanocomposites have been reported for high voltage direct current (HVDC) cable insulations, where ZnO nanoparticles suppress space charge accumulation in the insulating polymer matrix so that conductivity is reduced under high electric field.10 An increase in breakdown voltage was also achieved by decreasing the particle size from the μm to nm, which allows higher voltages in the cables.11The challenge in the development of the HVDC-insulation nanocomposites is to find high purity nanoparticles with narrow size distributions and low concentrations of conducting counter-ions on the particle surfaces. This is required not only for the electrical insulating characteristics but also for the predictable and successful application of uniform particle coatings that enable the dispersion of hydrophilic particles in hydrophobic polymers. Commercial ZnO nanoparticles are available, but the history of the particles is mostly unknown. Their purity is unspecified or not well documented, and micrographs reveal a mixture of morphologies with a large variation in particle size, (ESI Fig. S1†). Robust and reliable particle synthesis protocols are therefore needed, that allow the preparation of well-defined and high purity ZnO nanoparticles for use in polymer composites.
Various methods can be used to synthesize ZnO: hydrothermal (synthesis at temperature above 100 °C),12 vapour phase transport,13 solvent-based ultrasonic irradiation,14 microemulsion,15 aqueous sol–gel precipitation,16 and crystal growth on substrates.7,17,18 The flame and vapor-phase transport methods are used on a large scale, but with the drawbacks of mixed morphologies, particles sizes ranging up to micrometers, and impurities from the carbon catalysis of the ZnO gas phase growth.13,19,20 Microemulsion synthesis is efficient for obtaining specific particle sizes but the obtained amounts are small and expensive chemicals are used in addition to the intrinsic ZnO components. In contrast, the water-based chemistry is versatile and inexpensive, gives a high yield and can be up-scaled for continuous production.2 However, the heterogeneous nucleation related to the mixing/precipitation reactions lead to differences in particle size and morphology due to local variations in the particle growth conditions. These conditions are severely affected by the type of zinc metal salt used for the precipitation reaction since zinc salts with different counter-ions (chlorides, sulphates, acetates and nitrates) lead to different nucleation and growth kinetics. Their influence on the size, morphology, crystallographic and photoluminescence properties of the aqueous precipitate (ZnO) has been reported in separate studies for different precursors at different conditions,2,16,21–29 but have not been compared for a specific reaction condition yielding gram amounts of nanoparticle product. Another aspect of the salt selection is that the by-products show different behaviours when the suspensions are cooled to room temperature for cleaning of the particles. The sodium salt residuals after synthesis have different solubility, which also may affect the possibility of obtaining high purity ZnO nanoparticles.
This paper presents for the first time a comparison of different zinc salts used for high-yield aqueous synthesis of ZnO particles (∼10 g L−1) intended for high-voltage-insulation applications. The study compares the ZnO particle morphologies in relation to the zinc counter-ions used under identical synthesis conditions: Cl−, SO42−, NO3−, C2H3O2−. The results show that reproducible quantities of 8 g L−1 sub-50 nm ZnO nanoparticles with predictable surface characteristics can be obtained by careful selection of the zinc salt precursor. The ability of the different zinc salt precursor counter-ions to stabilize the primary nanoparticles during growth is discussed, and high-resolution crystallographic characterization and photoluminescence spectra are presented. An ultrasonication particle surface cleaning method is described, allowing for efficient removal of reaction by-products, wherein an induced particle aggregate fragmentation during cleaning resulted in two orders of magnitude smaller sized agglomerates. The reduced particle association resulted in stable colloidal suspensions of the particles that showed no evidence of sedimentation.
2. Experimental
2.1. Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, ≥98% (Sigma Aldrich)), zinc acetate dihydrate (Zn(CH3COO)2·2H2O, ≥99% (Sigma Aldrich)), zinc sulphate heptahydrate (ZnSO4·7H2O, ≥99.5% (Merck-KGaA)), zinc chloride (ZnCl2), ACS reag. grade, (Merck-KGaA), and sodium hydroxide (NaOH, ≥98%, (Sigma Aldrich)) were used as received. High resistivity MilliQ water, MilliQ: ‘ultrapure’ water of “Type 1”, following ISO 3696 and ASTM D1193-91 (18.2 Mohm cm at 25 °C) was used in all the reactions. Commercial ZnO nanoparticles; MK-ZnO-030 (MkNano, Canada) and ZnO-NanoTek® (Alfa Aesar, Germany) were purchased for comparison with the synthesized particles. They were specified to contain ZnO nanoparticles, 30 nm and 71 nm in size, respectively.2.2. Synthesis of zinc oxide nanoparticles
Table 1 summarizes the reaction conditions. The reactions were presumed to proceed via the following reactions:| Zn(X)2 + 2NaOH → ZnO + 2NaX + H2O | (1) |
| Zn(X) + 2NaOH → ZnO + Na2X + H2O | (2) |
| Sample | Metal salt precursor (aq.) | Metal salt [M] | NaOH [M] | Yield [g L−1] |
|---|---|---|---|---|
| ZN-8g | Zn(NO3)2 ·6H2O | 0.20 | 0.500 | 8 |
| ZC-8g | ZnCl2 (anhydrous) | 0.20 | 0.500 | 8 |
| ZS-8g | ZnSO4·7H2O | 0.20 | 0.500 | 8 |
| ZA-8g | Zn(O2CCH3)2·2H2O | 0.20 | 0.500 | 8 |
| ZA-2g | Zn(O2CCH3)2·2H2O | 0.05 | 0.125 | 2 |
| ZA-4g | Zn(O2CCH3)2·2H2O | 0.10 | 0.250 | 4 |
| ZA-6g | Zn(O2CCH3)2·2H2O | 0.15 | 0.375 | 6 |
| ZA-10g | Zn(O2CCH3)2·2H2O | 0.25 | 0.625 | 10 |
2.3. Nanoparticle cleaning and drying
After synthesis, the particle suspensions were transferred in 10 × 50 mL plastic centrifuge bottles and the reaction medium was replaced with the same volume of MilliQ water after each centrifugation, the only difference being that half of the suspended particles were exposed to ultrasonication as an intermediate treatment before the next centrifugation and water replacement. The ultrasonication treatment was performed for 15 min at 23 °C (Bandelin Sonorex RK 100H, volume = 3 L, ultrasonic peak output = 320 W, frequency = 35 kHz). The applied energy per volume (weight) of nanoparticle material in suspension was estimated to be ca. 4.8 kJ per 50 mL of particles (0.4 g). These samples are referred to as ultrasonically cleaned (UC). The suspensions not exposed to ultrasonication were intensively shaken by hand after water replacement and are referred to as traditionally washed (TW). Fig. 1 shows the flowchart of the two cleaning methods.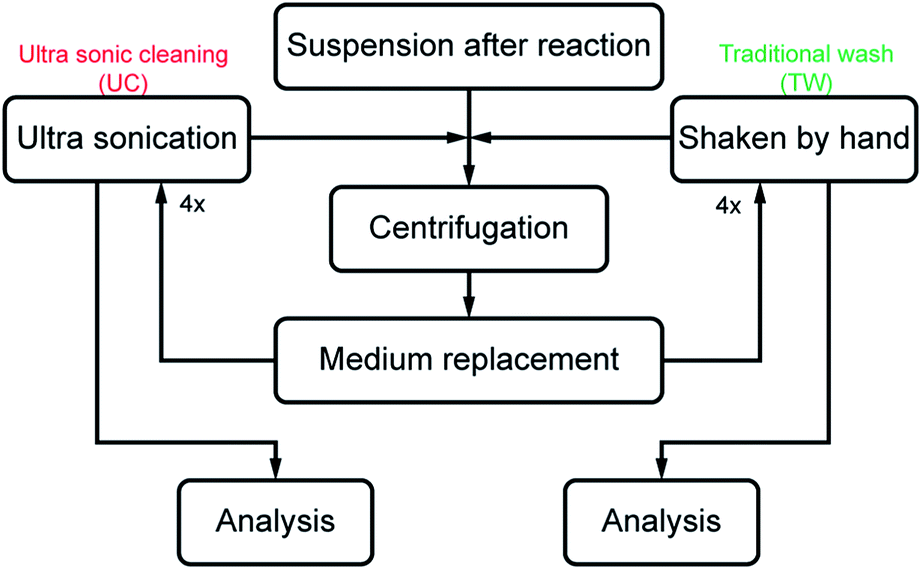 | ||
| Fig. 1 Experimental scheme for ultrasonic cleaning and traditional washing of synthesised ZnO nanoparticles. | ||
After the fourth cleaning cycle, particles were dried at 80 °C (air) and atmospheric pressure, followed by manual grinding to a fine powder with a pestle and mortar. The powders were further dried in a vacuum oven at 60 °C and 20 kPa for 2 h. Small samples (5 mL) were taken after each intermediate step in the cleaning procedure for characterization using dynamic light scattering (Section 2.4) in wet state.
2.4. Nanoparticle characterisation
Field emission scanning electron microscopy (SEM; Hitachi S-4800) and transmission electron microscopy (TEM; Hitachi HT7700) were used to determine the size and volume distributions of the ZnO particles. For SEM: powder samples were coated in a Cressington 208 HR with a thin conductive layer of Pt/Pd (60/40) by sputtering for 20 s at 80 mA. Examination was carried out at an acceleration voltage of 5 kV and a current of 10 μA. For TEM: the particles were deposited on 400 mesh copper grids, with an ultrathin holey/lacey carbon film (product no. 01824, Ted Pella, Inc. USA), from an ultrasonicated suspension of pure ethanol containing a particle concentration of 0.45 ± 0.05 g L−1. The prepared samples were dried and examined at 100 kV acceleration voltage. The size distributions of the nanoparticles were obtained by manually measuring 600 particles (for each sample) using ImageJ (National Institute of Health, Maryland, USA), and the volume distributions were calculated from the radii of the individual particles.X-ray diffractograms of the powder samples were taken at room temperature using a PANalytical X'pert Pro MPD diffractometer with a Cu-Kα source (wavelength 1.54178 Å) at a step size of 0.017° (2θ).
Dynamic light scattering (DLS) was used to assess the average aggregate size of the particles with simultaneous suspensions electrical conductivity (mS cm−1) and zeta potential measurements using a Malvern Zetasizer Nano ZS (Malvern, UK). Measurements were performed with a 633 nm He/Ne laser at a temperature of 25 °C for dispersed particles in water with different concentrations (2 to 10 g L−1). The refractive index (RI) of ZnO particles was set to 2.004. Prior to the size and conductivity measurements, the UC suspensions were ultrasonicated for 15 min and TW samples were shaken by hand. The pH of the suspensions was measured by pH 700 (Eutech Instruments, Singapore). All these samples were maintained as wet from the synthesis.
Room temperature photoluminescence (PL) emission measurements were made in a Perkin Elmer LS55 instrument with Xe lamp. 1 mL of UC suspensions (ZN-8g, ZC-8g, ZS-8g, and ZA-8g) were diluted with 2 mL water and placed in a quartz cuvette with an inner cross section of 10 × 10 mm2. The excitation wavelength was 290 nm, and both excitation and emission slit widths were 5 nm.
3. Results and discussion
3.1. Morphology of synthesized nanoparticles
Fig. 2a–d present electron micrographs of the nanoparticles synthesized in 8 g batches (Table 1), from the different zinc salts. The most narrowly sized particles with an average size of 25 nm were dominantly of cone-shaped prism morphology (>60%) and obtained from the acetate precursor (Fig. 2a, size distributions: Fig. 3a). The chloride and sulphate precursors yielded both 10–30 nm sized particles and significantly larger particles (petals) with an average size of 80–100 nm (Fig. 2b and c). In the case of the nitrate precursor, the petals condensed into larger, star-shaped particles with an average size of ca. 500 nm (Fig. 2d). Klaumünzer et al.30 suggested that these petals stemmed from an oriented self-assembly and condensation of the nanoprisms (along the c-axis [0001/000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ]), which herein reached up to ca. 300 nm in size, i.e. 10 times larger than the nanoprisms (size and volume distribution Fig. 3a). The petals (Fig. 2b and c) in a second step formed octahedrons (twins of petals) that interconnected via condensation of the wider base of the prisms/petals, in accordance with reports by Yang and Xie et al.30–32 Two octahedron shaped polygons are depicted in the top right-hand corner of Fig. 2c (representing <1% amount of particles, Fig. 3a) and as separately synthesized in the ESI (Fig. S2†). Oliveira et al. proposed a similar reaction scheme and suggested that half the octahedrons (petals) were formed via aggregation of nanoprisms, and that the second half was then germinated on the wider base of the formed petal.33 Both schemes are plausible, although in the present work, the micrographs showed the fusion of equally large petals was dominant. The larger octahedrons showed at different stages during the growth sufficient local surface energy to allow additional petals to germinate from the centre of the octahedron, transforming the two-sided cones into ca. 500 nm uniform stars (Fig. 2d, inset), also referred to as flowers.16 A micrograph showing this intermediate stage before the fully developed flower shapes is included in the ESI (Fig. S3†).
]), which herein reached up to ca. 300 nm in size, i.e. 10 times larger than the nanoprisms (size and volume distribution Fig. 3a). The petals (Fig. 2b and c) in a second step formed octahedrons (twins of petals) that interconnected via condensation of the wider base of the prisms/petals, in accordance with reports by Yang and Xie et al.30–32 Two octahedron shaped polygons are depicted in the top right-hand corner of Fig. 2c (representing <1% amount of particles, Fig. 3a) and as separately synthesized in the ESI (Fig. S2†). Oliveira et al. proposed a similar reaction scheme and suggested that half the octahedrons (petals) were formed via aggregation of nanoprisms, and that the second half was then germinated on the wider base of the formed petal.33 Both schemes are plausible, although in the present work, the micrographs showed the fusion of equally large petals was dominant. The larger octahedrons showed at different stages during the growth sufficient local surface energy to allow additional petals to germinate from the centre of the octahedron, transforming the two-sided cones into ca. 500 nm uniform stars (Fig. 2d, inset), also referred to as flowers.16 A micrograph showing this intermediate stage before the fully developed flower shapes is included in the ESI (Fig. S3†).
 | ||
| Fig. 2 Morphologies of ZnO particles formed from: (a) zinc acetate (b) zinc chloride; (c) zinc sulphate and (d) zinc nitrate, prepared under identical conditions as 8 g batches. (e) and (f) top and side views of acetate-derived particles, respectively. The single crystal prisms are visible in (a), (e) and (f), arrows marked “1” show the multicrystal petals grown from prisms (b and c), and the arrow marked “2” shows the octahedrons from merged petals (c). | ||
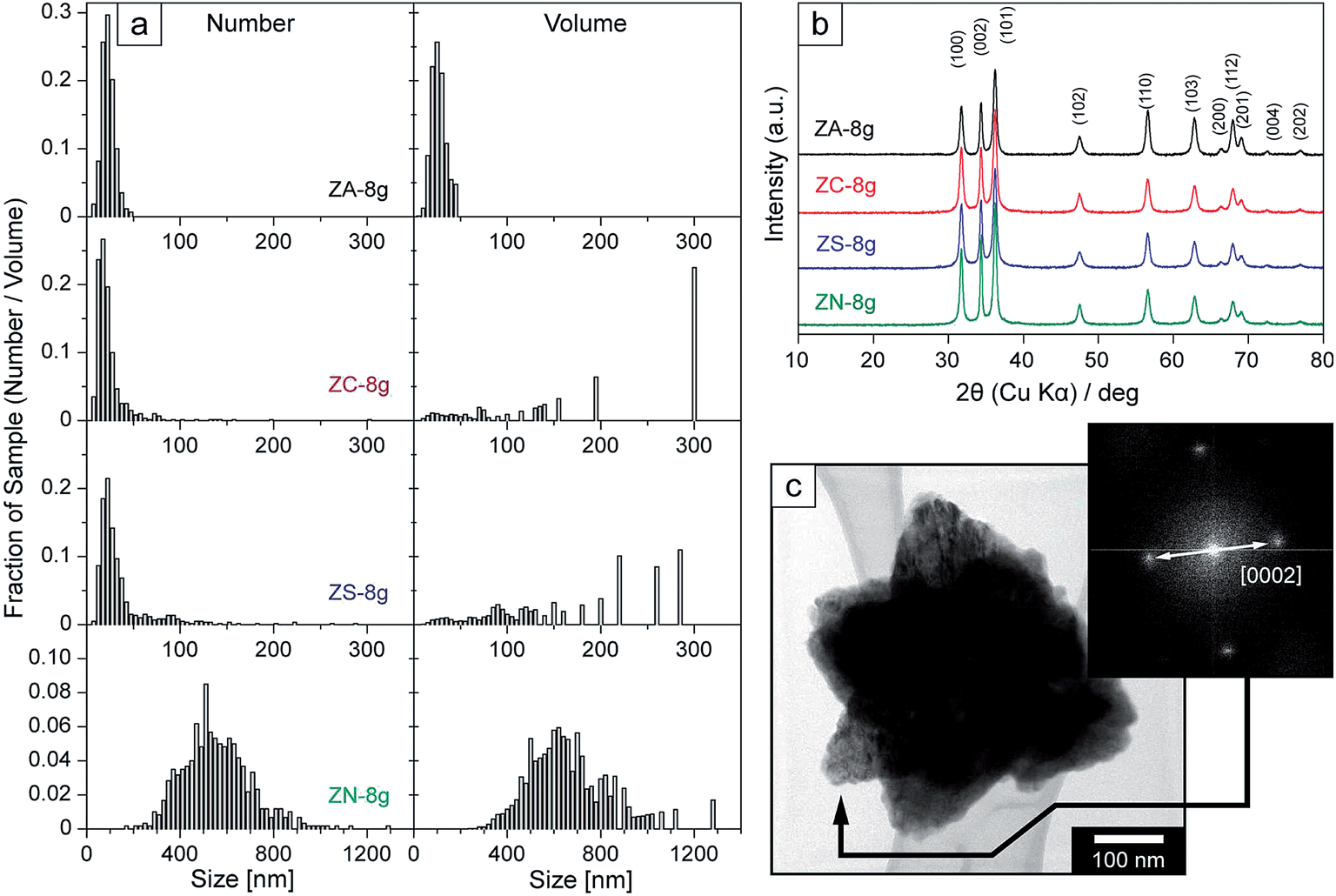 | ||
| Fig. 3 (a) (left) Relative frequency distribution of particle size; (right) relative volume frequency distribution of particle size; (b) X-ray diffractograms for 8 g batches synthesized from different zinc salts; (c) TEM of flower-shaped particle with diffraction pattern taken at point of arrow. | ||
The prisms consequently grew into larger structures in the chloride, sulphate and nitrate cases by migration of prisms and addition of matter added via dissolution and re-precipitation of material “sealing” the prisms into uniform petals, octahedrons and flower shapes.34,35 Fig. 2e and f show high resolution TEM of the nanoprisms from the acetate salt with step edged surfaces along the prism side (the preferred c-axis growth direction). Joo et al.36 attributed the step edge creation to a higher growth rate in the [0001] direction than in the perpendicular direction. In conclusion, the cause of the variation in morphology is the different abilities of the counter-ions to electrostatically stabilize individual nanoprisms into isolated highly crystalline solids that remain isolated during the full course of the reaction (see Section 3.3). The strong ability of the acetate ions to favour this stabilization has been suggested to originate from strong uni- and bi-dentate oxygen coordination bonding of the acetate ions to individual zinc atoms, or parallel bridging of the two oxygen atoms in the acetate ions37–41 to positively charged zinc atoms in the Zn enriched surface planes {101} of the ZnO particles.31,42,43 However, the dominant state of the deposited zinc hydroxide precursor are Zn(OH)3− and Zn(OH)42− species, which initially should cause negatively charged surface during the crystal growth in the aqueous medium.44 It is therefore suggested that the stabilization of individually growing nanoprisms possibly resulted from a formed amphiphilic capping layer around the nanoprisms, where the partially positively charged methyl functional unit of the acetate ions was associated with the insufficiently condensed negatively charged zinc hydroxide structures reported by Nicholas et al.45 (see Section 3.2). This formed capping layer potentially prevented extensive fusion of the particles in a similar fashion as citrates are used to during controlled growth of complex and oriented ZnO structures.17 This reasoning would be consistent with the absence of any carbon functional units in the chloride, sulphate and nitrate cases and their significantly smaller coordination capacity as compared to the acetate counter-ions. The difference between the anions (Cl−, SO42−, NO3−) may also relate to their different interactions with the sodium ion (Na+) by-product from the reactions, see Section 3.3.
3.2. Size/volume distributions and crystal structure
Fig. 3a (left) shows the distribution of the particles cross-sectional diameter, while Fig. 3a (right) shows how much of the volume of the entire samples the differently sized particles constituted. It should be noted that a few larger particles obtained from the chloride and sulphate salts had a marked impact on the volume distributions of the particles. In Fig. 3a (right) it can be seen that almost 50 vol% of the precipitated materials is related to particle sizes greater than 100 nm, and that 23 vol% of the material in the chloride suspensions is related to the ca. 300 nm petals.Fig. 3b shows the X-ray diffractograms of the samples produced from different zinc salt precursors. All the samples showed a single phase ZnO with clear diffraction peaks corresponding to the lattice planes in the wurzite with a hexagonal cell structure, and crystallographic parameters of a = b = 3.254 Å, c = 5.210 Å, α = β = 90°; γ = 120° (inorganic crystal structure database; collection code # 067849). The unit cell of a mono-domain particle with the interplanar spacing of 2.6 Å (half size of unit cell in c-direction) is shown in Fig. 2f (the lattice also being confirmed from the basal plane of the prisms, Fig. S4 (ESI†) as 2.8 Å). The absence of an amorphous halo and the sharpness of the peaks characteristic of ZnO indicated high crystallinity and high purity for all the particles.
The crystal size was obtained from the Scherrer equation:
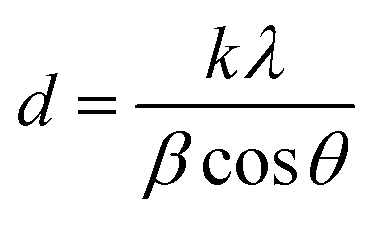 | (3) |
3.3. Characteristics of residues in the suspensions immediately after the ZnO synthesis
Fig. 4 shows the ZnO particles formed from the different precursors together with their reaction residuals, taken as aliquots immediately from the reaction suspensions (without cleaning). The different precursors generated non-ZnO phases which greatly differed both in terms of their association with the surface of the ZnO particles and/or their preference for the TEM grid surface. In all the suspensions, the residuals were present as highly hydrated salts with a bound water phase that evaporated as it was extensively exposed to the electron beam (>15 seconds) in the microscope. The residuals are referred to as hydrated salts of sodium (from the alkaline NaOH used to enforce the precipitations) with the respective counter-ions from the zinc source. All show lower solubility limits (20 °C) of their corresponding salts (NaCH3COO: 464 g L−1; NaCl: 359 g L−1; Na2SO4: 195 g L−1 and NaNO3: 876 g L−1) as compared to NaOH: 1090 g L−1,47 which was also consumed during the ZnO precipitation (reaction schemes 1 and 2). | ||
| Fig. 4 TEM micrographs (a) to (d) the preference of salt residuals to the ZnO particle surfaces observed when immediately deposited on TEM grids from reaction suspensions; (a) acetate; (b) chloride; (c) sulphate; (d) nitrate. The arrows point to the salt residuals. SEM micrographs (e) and (f) show the effect of decreasing the concentration of precursor on particle morphologies for chloride and acetate precursors, respectively. | ||
Fig. 4a shows that the salt residuals from the acetate suspension showed the strongest preference for the particle surfaces of all the samples and displayed 15–20 nm covers on and between the particles. A small amount was also spread evenly on the carbon surface of the grids as a cracked layer (Fig. 4a). The chloride-based residuals appeared as separate phase from the particles in lumps ca. 100–500 nm in size along the Formvar (polyvinyl formal polymer) borders (Fig. 4b), but they were also found as a ca. 5–10 nm covers on the particles (Fig. 4b inset). The sulphate residuals were almost uniquely precipitated as uniformly shaped ovals along the Formvar edges (Fig. 4c) and showed significantly less attraction to the surface of the ZnO crystals. Fig. 4d shows that the sodium nitrate residue appeared as 20–40 nm facetted solids with sharp edges being markedly different from the other salt residuals. These facetted solids were distributed evenly on the surface of the Formvar polymer (Fig. 4d) and no salt covers could be observed on the edges of the crystals. This “sharp-edged” material was transformed into a uniform and almost transparent layer of dehydrated salt upon exposure to the electron beam (ESI Video 1†) and it was unclear whether the facetted residuals precipitated in parallel with the ZnO precipitation or formed on the TEM grid after synthesis. It is suggested the different shape of facetted residuals led a more extensive migration of the primary nanoprisms into the petal shapes that developed the submicron flower shapes. The opposite behaviour was suggested for the acetate counter-ions that stabilized the individual nanoprisms during the crystal growth and prevented their assembly to larger petals.
Fig. 4e and f compare the morphologies of the formed particles from chloride and acetate systems when the amount of counter-ions per reaction suspension volume was reduced by 50% (reaction schemes 1 and 2) to investigate a possible “shielding effect” of different counter-ions on the nanoprisms stabilisation. The reduced concentration of the chloride species resulted in strongly enhanced primary nanoprisms associations and a semi-complete formation of flower-shaped units similar to those derived from the nitrate solutions (Fig. 4e and 2d). The inability of the nitrate counter-ions to stabilize the nanoprisms was also experimentally confirmed by reducing precursor concentration which resulted in four-fold increase in size of flower-shaped particles to ca. 2 μm with the growth of additional petals (ESI Fig. S5†). Interestingly, advantage has been taken of the low ability of the nitrate-based precursor to restrict the migration and association of smaller particles in several scientific studies aimed to grow specific directional morphologies in hydroxide solutions.16–18,48–50 The growth kinetics and particle size variation were previously related to “virtual” capping shells created by the cations of the alkaline source (NaOH, LiOH or KOH).51,52 It is here emphasized that selection of zinc salt precursor appears to have a more significant effect on the outcome of the reactions under otherwise identical precipitation conditions. It could also be concluded that only the acetate ions had a strong ability to stabilize the primary nanoparticles as separate nanoprisms during the reaction, and this ability was generic for all reaction yields from 2 to 10 g L−1 (Table 1) since all gave solitary individual nanoparticles in the sub-50 nm regime.
3.4. Removal of supernatant impurities (reaction residuals)
Fig. 5 presents a typical sequence showing of how the hydrated sodium salts associate and adhere to the surface of the nanoparticles upon drying under the electron beam. The micrographs are taken after 0, 60, 90 and 120 seconds of exposure to a beam on an area with a significant amount of hydrated sodium sulphate residuals covering an agglomerate of nanoparticles. A complete video of the full sequence is available in the ESI, Video 2.† From the sequence of micrographs it can be concluded that the sodium salt supernatant did not evaporate evenly but instead contracted as droplets and condensed directly onto the surface of the particles as a coating. It is therefore of fundamental importance to properly remove the reaction residues in the wet state prior to any sort of drying, in order to avoid thin deposits of salt residuals.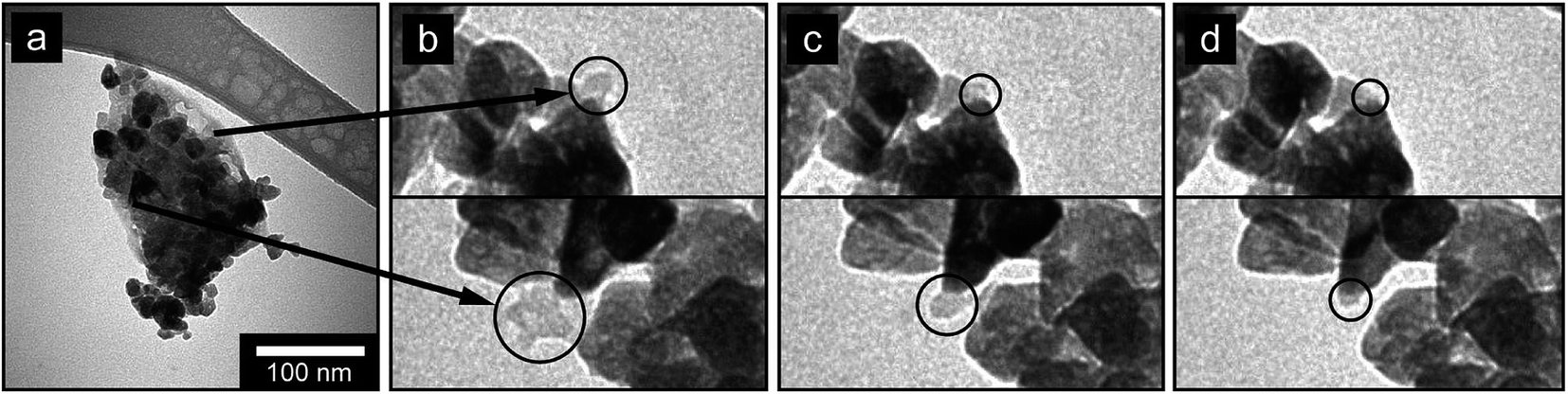 | ||
| Fig. 5 Micrographs (TEM) showing association and evaporation of salt supernatant with preference to the surface of the nanoparticles after (a) 0 s, (b) 60 s, (c) 90 s, (d) 120 seconds exposure to electron beam. | ||
Fig. 6a (inset) shows that the conductivity for different yields of the acetate-derived particles, which was proportional to the concentration of particles and remaining precursor counter-ions after the reaction. The acetate system was selected due to its preferential association to the particle surfaces (Fig. 4a, inset) and its ability to form equally uniform nanoparticles (25 ± 10 nm) with a large surface area for different reaction yields, see Fig. 4f. Accordingly, the suspension conductivity was here used as representative parameter of the residual counter-ions. Different methods have been proposed for removal of reaction residuals including solvent cleaning by hexane, heptane and methanol.37,38,53 Here, the efficiency of ultrasonic cleaning (UC) of the particles was compared with the more traditional repeated replacement of reaction supernatant with high purity water (TW), Fig. 1. Fig. 6a (top and bottom) show the electrical conductivity of the UC and TW suspensions against the number of cleaning cycles. The conductivity of the suspensions decreased by 1.5–2 orders of magnitude after the first cycle, followed by a decrease of one order of magnitude after the second cycle, regardless of the cleaning method. The overall decrease in suspension conductivity was about 3.5 orders of magnitude. There was slightly less scatter in the data for the UC method, but at the lower conductivities (after several cycles of cleaning), the conductivity approached that of MilliQ water and the precision decreased.
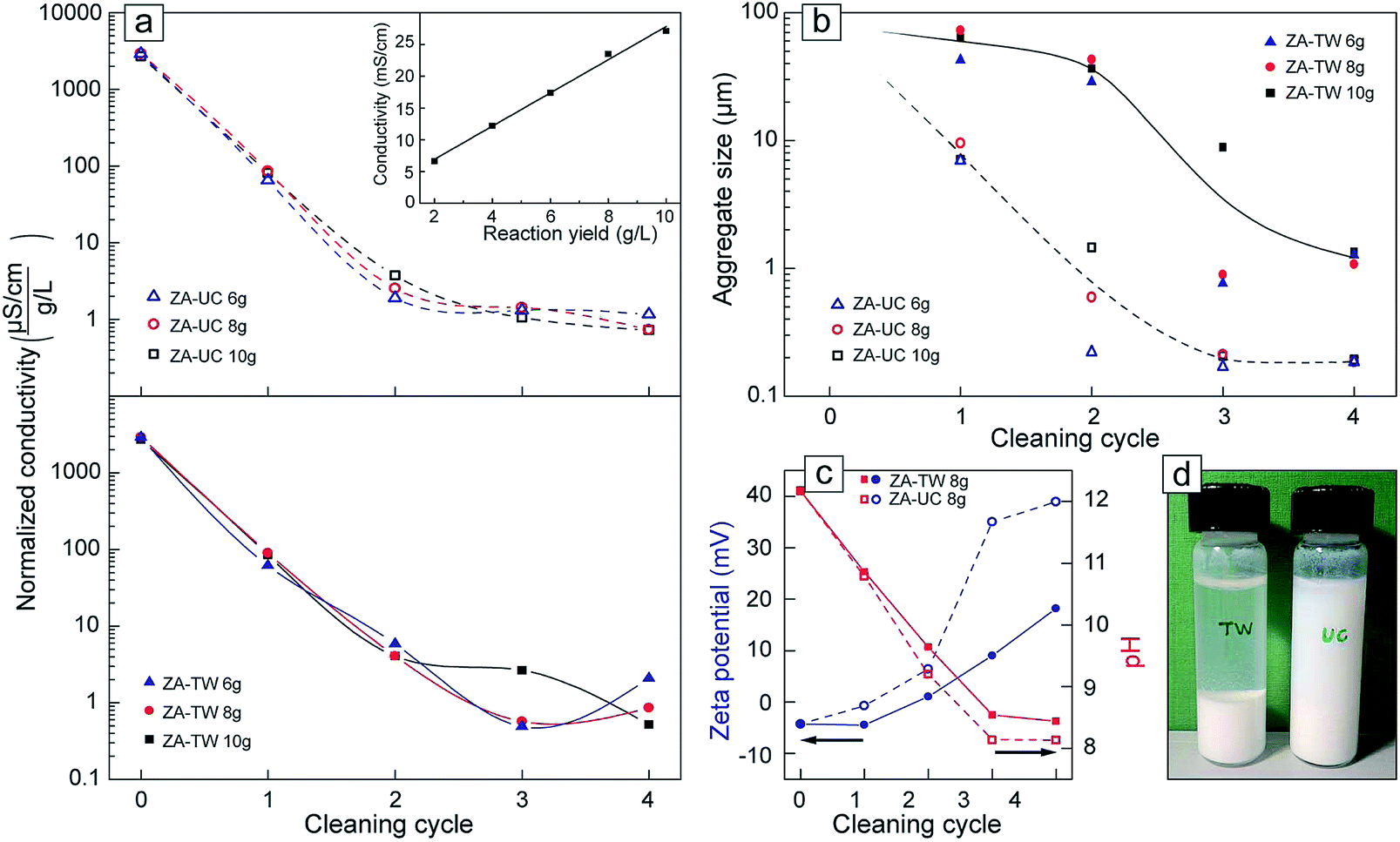 | ||
| Fig. 6 (a) The conductivity of the particle suspensions (normalized to with respect to yields): prior to, during and after particle cleaning (at fourth cycle). The top graph shows the UC (ultrasonic cleaning) and the bottom graph the TW (traditional washing) method. The inset in top right corner shows the linear correlation between conductivity and precipitate concentration. (b) The hydrodynamic size of the aggregates in the suspensions from DLS measurements. (c) Zeta potential and pH as a function of washing cycles, (d) photograph of sedimentation characteristic of TW and UC cleaned particles after 48 h. | ||
Fig. 6b shows the average size of the aggregates present after each cleaning cycle for UC and TW methods. In contrast to the conductivity measurements, the data showed a difference between the cleaning methods; the UC treatment resulted in aggregate sizes about one order of magnitude smaller those given by traditional washing (after the 4th cycle). The average cluster sizes of the ultrasonicated samples was ca. 200 nm for the more concentrated samples (8 g and 10 g), whereas the traditionally washed samples showed cluster sizes approximately 8 times larger; >1.5 μm for the same samples. From a practical perspective, the 8 g batches could be characterized by particle aggregates with an average of ca. 350 associated nanoparticles in the UC samples, whereas in the TW samples the aggregates consisted of approximately 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 particles (assuming tightly packed particles in both cases). It is here suggested that the large clusters/aggregates observed in the TW samples could be attributed to the precursor counter-ion from the synthesis, which remained in the interior of the traditionally washed particle clusters. The zeta potential of the UC samples after the last cycle also showed a significantly higher potential than the samples cleaned by traditional washing (Fig. 6c, inset). It is suggested that the ultrasonication method leads to more effective cleaning because of an evenly induced fragmentation of particle aggregates between every cleaning step,54 which leads to a more effective release of entrapped precursor salt counter-ions. This would also explain the less predictable conductivity data in the cycles using the regular (TW) method, where aggregates broke more sporadically as the energy provided by shaking was not always sufficient in the cleaning cycles (Fig. 6a). In terms of practical relevance, the higher zeta potential values (>+30 mV) in the UC samples were reflected in a greater colloidal stability of the suspensions. Samples cleaned by ultrasonication were completely stable and still suspended after 48 hours due to electrostatic repulsion between the particles, whereas the particles completely sedimented in the TW samples (Fig. 6d). The chloride- and sulphate-derived particle samples were characterized by EDX in the high resolution TEM and it was confirmed that there were no detectable traces of counter-ions remaining on their surfaces after UC cleaning (ESI Fig. S6†).
000 particles (assuming tightly packed particles in both cases). It is here suggested that the large clusters/aggregates observed in the TW samples could be attributed to the precursor counter-ion from the synthesis, which remained in the interior of the traditionally washed particle clusters. The zeta potential of the UC samples after the last cycle also showed a significantly higher potential than the samples cleaned by traditional washing (Fig. 6c, inset). It is suggested that the ultrasonication method leads to more effective cleaning because of an evenly induced fragmentation of particle aggregates between every cleaning step,54 which leads to a more effective release of entrapped precursor salt counter-ions. This would also explain the less predictable conductivity data in the cycles using the regular (TW) method, where aggregates broke more sporadically as the energy provided by shaking was not always sufficient in the cleaning cycles (Fig. 6a). In terms of practical relevance, the higher zeta potential values (>+30 mV) in the UC samples were reflected in a greater colloidal stability of the suspensions. Samples cleaned by ultrasonication were completely stable and still suspended after 48 hours due to electrostatic repulsion between the particles, whereas the particles completely sedimented in the TW samples (Fig. 6d). The chloride- and sulphate-derived particle samples were characterized by EDX in the high resolution TEM and it was confirmed that there were no detectable traces of counter-ions remaining on their surfaces after UC cleaning (ESI Fig. S6†).
3.5. Photoluminescence of the ZnO particles
All the samples showed a broad emission ranging from 368 to 645 nm with the emphasis in the blue-violet region. The spectra were deconvoluted into a series of four relatively sharp Gaussian peaks at 417 nm (2.97 eV), 459 nm (2.70 eV), 484 nm (2.56 eV) and 530 nm (2.34 eV) and a less sharp peak at 405 nm (3.06 eV), Fig. 7.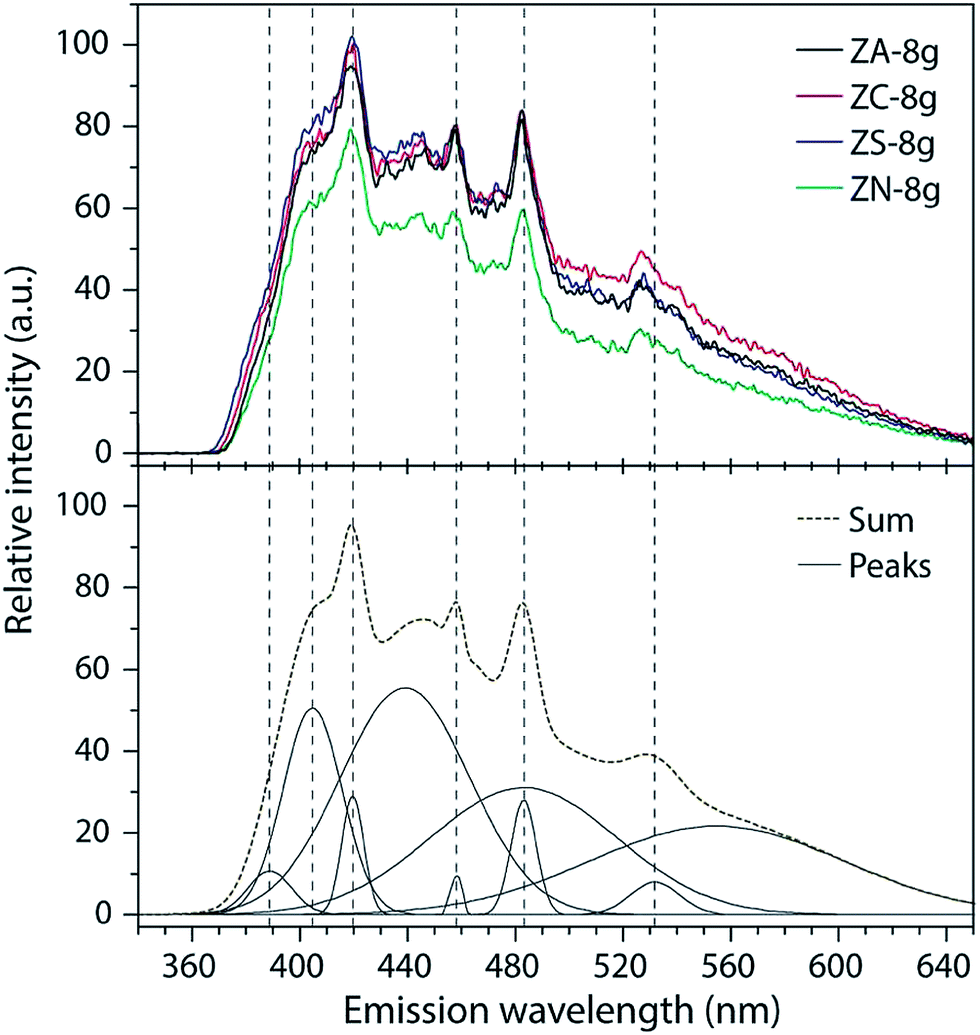 | ||
| Fig. 7 (top) PL spectra of ZnO particles synthesized from different zinc salt precursors measured at room temperature with (bottom) deconvoluted peaks. The excitation wavelength is 290 nm. | ||
Photoluminescence (PL) spectra of ZnO have been explained as being due to various mechanisms with the recombination from excitation giving the peak of shortest wavelength at 386 nm (3.21 eV).55 According to the literature there are many energy levels due to various defects such as zinc and oxygen vacancies and/or interstitials.56 Ye et al.57 suggest that the green emission relates to transitions from a conduction band to oxygen vacancies at ca. 500 nm in the grain boundary, and to transitions from oxygen vacancies to the valence band at ca. 560 nm in the bulk grain. These peaks were responsible for the broad response in Fig. 7. The relatively sharp peak at 530 nm (also visible in Fig. 7) has been reported by Wu et al.58 Zeng et al.59 reported blue peaks at 415 nm, 440 nm, 455 nm and 488 nm. These correspond well with our spectra, although the preparation method was vastly different, i.e. pulsed laser deposition. Overall, the ZnO particles derived from different precursors showed almost identical spectra, the only difference being that nitrate-derived particles showed ca. 20% less intensity, which is probably due to the larger particle size. Since PL properties of ZnO in general are very sensitive to defects, it is concluded that the 25 nm prism formations were to a high degree unaffected by the precursor type, regardless of the final particle morphology.
4. Conclusions
The successful high-yield (>8 g L−1) aqueous precipitation synthesis of zinc oxide nanoparticles with narrow size distribution was critically dependent on the selection of zinc salt precursor. Among the C2H3O2−, Cl−, SO42− and NO3− counter-ions, only acetate resulted solely in 25 nm nanoparticles with a narrow size distribution due to its strong ability to stabilize the primary nanoparticles. On the other hand, the inability of the nitrate precursor to stabilize the nanoprisms allowed extensive self assembly along the c-axis of the nanoprism into petals and octahedrons, which via further germination developed into spiky flower shapes. Chloride and sulphate counterions showed an intermediate behaviour resulting in mixed morphologies. However, regardless of the morphology, the specific photoluminescence properties of the different sized particles showed identical emission ranging from 368 to 645 nm with an emphasis in the blue-violet regions. This suggests that the interlayered phases that developed during the assembly of the nanoprisms have no significant effect on the photoluminescence characteristics of the ultimate particles. Finally, ultrasonic particle surface cleaning compared to water replacement showed a more efficient removal of counter-ions from the particles, resulting in colloidal stability of ZnO nanoparticles even after 48 h due to electrostatic stabilization of the particles.Acknowledgements
The financial support from the Swedish Foundation for Strategic Research (SSF, Ref. EM11-0022) is gratefully acknowledged.References
- A. B. Djurisic, X. Chen, Y. H. Leung and A. M. Ching Ng, J. Mater. Chem., 2012, 22, 6526–6535 RSC.
- L. Wang and M. Muhammed, J. Mater. Chem., 1999, 9, 2871–2878 RSC.
- J. K. Cooper, Y. Ling, C. Longo, Y. Li and J. Z. Zhang, J. Phys. Chem. C, 2012, 116, 17360–17368 CAS.
- M. Kevin, W. H. Tho and G. W. Ho, J. Mater. Chem., 2012, 22, 16442–16447 RSC.
- G. Wrobel, M. Piech, P. X. Gao and S. Dardona, Cryst. Growth Des., 2012, 12, 5051–5055 CAS.
- S. Xu, Y. Shen, Y. Ding and Z. L. Wang, Adv. Funct. Mater., 2010, 20, 1493–1497 CrossRef CAS PubMed.
- R. Kozhummal, Y. Yang, F. Güder, A. Hartel, X. Lu, U. M. Küçükbayrak, A. Mateo-Alonso, M. Elwenspoek and M. Zacharias, ACS Nano, 2012, 6, 7133–7141 CrossRef CAS PubMed.
- Dielectric Polymer Nanocomposites, ed. J. K. Nelson, Springer, New York, 2010 Search PubMed.
- S. Sahoo, M. Maiti, A. Ganguly, J. J. George and A. K. Bhowmick, J. Appl. Polym. Sci., 2007, 105, 2407–2415 CrossRef CAS PubMed.
- F. Tian, Q. Lei, X. Wang and Y. Wang, Appl. Phys. Lett., 2011, 99, 142903 CrossRef PubMed.
- J. I. Hong, L. S. Schadler, R. W. Siegel and E. Martensson, Appl. Phys. Lett., 2003, 82, 1956–1958 CrossRef CAS PubMed.
- M. Søndergaard, E. D. Bøjesen, M. Christensen and B. B. Iversen, Cryst. Growth Des., 2011, 11, 4027–4033 Search PubMed.
- S. Polarz, A. Roy, M. Merz, S. Halm, D. Schröder, L. Schneider, G. Bacher, F. E. Kruis and M. Driess, Small, 2005, 1, 540–552 CrossRef CAS PubMed.
- G. Applerot, A. Lipovsky, R. Dror, N. Perkas, Y. Nitzan, R. Lubart and A. Gedanken, Adv. Funct. Mater., 2009, 19, 842–852 CrossRef CAS PubMed.
- C. Lu and C. Yeh, Mater. Lett., 1997, 33, 129–132 CrossRef CAS.
- S. Sepulveda-Guzman, B. Reeja-Jayan, E. de la Rosa, A. Torres-Castro, V. Gonzalez-Gonzalez and M. Jose-Yacaman, Mater. Chem. Phys., 2009, 115, 172–178 CrossRef CAS PubMed.
- Z. R. Tian, J. A. Voigt, J. Liu, B. Mckenzie, M. J. Mcdermott, M. A. Rodriguez, H. Konishi and H. Xu, Nat. Mater., 2003, 2, 821–826 CrossRef CAS PubMed.
- Z. Wang, X. Qian, J. Yin and Z. Zhu, Langmuir, 2004, 20, 3441–3448 CrossRef CAS.
- C. Lin, C. Hwang, W. Lee and W. Tong, Mater. Sci. Eng., B, 2007, 140, 31–37 CrossRef CAS PubMed.
- D. Banerjee, J. Y. Lao, D. Z. Wang, J. Y. Huang, Z. F. Ren, D. Steeves, B. Kimball and M. Sennett, Appl. Phys. Lett., 2003, 83, 2061–2063 CrossRef CAS PubMed.
- C. Wu, X. Qiao, J. Chen, H. Wang, F. Tan and S. Li, Mater. Lett., 2006, 60, 1828–1832 CrossRef CAS PubMed.
- N. Goswami, Advances in Synthesis, Processing, and Applications of Nanostructures, Ceram. Trans., 2012, 238, 3–15 CAS.
- J. Lee, A. J. Easteal, U. Pal and D. Bhattacharyya, Curr. Appl. Phys., 2009, 9, 792–796 CrossRef PubMed.
- N. Goswami and D. K. Sharma, Physica E, 2010, 42, 1675–1682 CrossRef CAS PubMed.
- A. Moezzi, M. Cortie and A. McDonagh, Dalton Trans., 2011, 40, 4871–4878 RSC.
- T. Kawano and H. Imai, Colloids Surf., A, 2008, 319, 130–135 CrossRef CAS PubMed.
- T. Kawano and H. Imai, Cryst. Growth Des., 2006, 6, 1054–1056 CAS.
- B. Li and Y. Wang, J. Phys. Chem. C, 2010, 114, 890–896 CAS.
- A. Becheri, M. Dürr, P. L. Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10, 679–689 CrossRef CAS.
- M. Klaumünzer, M. Distaso, J. Hübner, M. Mačković, E. Spiecker, C. Kryschi and W. Peukert, CrystEngComm, 2014, 16, 1502–1513 RSC.
- M. Yang, K. Sun and N. A. Kotov, J. Am. Chem. Soc., 2010, 132, 1860–1872 CrossRef CAS PubMed.
- R. Xie, D. Li, H. Zhang, D. Yang, M. Jiang, T. Sekiguchi, B. Liu and Y. Bando, J. Phys. Chem. B, 2006, 110, 19147–19153 CrossRef CAS PubMed.
- A. P. A. Oliveira, J.-F. Hochepied, F. Grillon and M.-H. Berger, Chem. Mater., 2003, 15, 3202–3207 CrossRef CAS.
- P. Uthirakumar, B. Karunagaran, S. Nagarajan, E. Suh and C. Hong, J. Cryst. Growth, 2007, 304, 150–157 CrossRef CAS PubMed.
- S. Musić, Đ. Dragčević and S. Popović, J. Alloys Compd., 2007, 429, 242–249 CrossRef PubMed.
- J. Joo, S. G. Kwon, J. H. Yu and T. Hyeon, Adv. Mater., 2005, 17, 1873–1877 CrossRef CAS PubMed.
- D. Sun, M. Wong, L. Sun, Y. Li, N. Miyatake and H. J. Sue, J. Sol-Gel Sci. Technol., 2007, 43, 237–243 CrossRef CAS.
- D. Segets, R. Marczak, S. Schäfer, C. Paula, J. Gnichwitz, A. Hirsch and W. Peukert, ACS Nano, 2011, 5, 4658–4669 CrossRef CAS PubMed.
- S. Sakohara, L. D. Tickanen and M. A. Anderson, J. Phys. Chem., 1992, 96, 11086–11091 CrossRef CAS.
- S. Sakohara, M. Ishida and M. A. Anderson, J. Phys. Chem. B, 1998, 102, 10169–10175 CrossRef CAS.
- R. D. Yang, S. Tripathy, Y. Li and H. J. Sue, Chem. Phys. Lett., 2005, 411, 150–154 CrossRef CAS PubMed.
- X. Liu and M. T. Swihart, Nanoscale, 2013, 5, 8029–8036 RSC.
- B. Ludi and M. Niederberger, Dalton Trans., 2013, 42, 12554–12568 RSC.
- A. Degen and M. Kosec, J. Eur. Ceram. Soc., 2000, 20, 667–673 CrossRef CAS.
- N. J. Nicholas, G. V. Franks and W. A. Ducker, Langmuir, 2012, 28, 7189–7196 CrossRef CAS PubMed.
- S. Liu, E. Killen, M. Lim, C. Gunawan and R. Amal, RSC Adv., 2014, 4, 4363–4370 RSC.
- A. Seidell, Solubilities of inorganic and metal organic compounds, D. Van Nostrand Company, New York, 3rd edn, 1940, vol. 1 Search PubMed.
- S. Cho, S. Jung and K. Lee, J. Phys. Chem. C, 2008, 112, 12769–12776 CAS.
- F. Xu, Y. Lu, Y. Xie and Y. Liu, J. Phys. Chem. C, 2009, 113, 1052–1059 CAS.
- T. L. Sounart, J. Liu, J. A. Voigt, J. W. P. Hsu, E. D. Spoerke, Z. Tian and Y. B. Jiang, Adv. Funct. Mater., 2006, 16, 335–344 CrossRef CAS PubMed.
- R. Viswanatha, H. Amenitsch and D. D. Sarma, J. Am. Chem. Soc., 2007, 129, 4470–4475 CrossRef CAS PubMed.
- P. K. Santra, S. Mukherjee and D. D. Sarma, J. Phys. Chem. C, 2010, 114, 22113–22118 CAS.
- E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566–5572 CrossRef CAS.
- S. J. Chung, J. P. Leonard, I. Nettleship, J. K. Lee, Y. Soong, D. V. Martello and M. K. Chyu, Powder Technol., 2009, 194, 75–80 CrossRef CAS PubMed.
- J. J. Wu and S. C. Liu, Adv. Mater., 2002, 14, 215–218 CrossRef CAS.
- S. Vempati, J. Mitra and P. Dawson, Nanoscale Res. Lett., 2012, 7, 470 CrossRef PubMed.
- J. D. Ye, S. L. Gu, F. Qin, S. M. Zhu, S. M. Liu, X. Zhou, W. Liu, L. Q. Hu, R. Zhang, Y. Shi and Y. D. Zheng, Appl. Phys. A, 2005, 81, 759–762 CrossRef CAS.
- D. Wu, Z. Huang, G. Yin, Y. Yao, X. Liao, D. Han, X. Huang and J. Gu, CrystEngComm, 2010, 12, 192–198 RSC.
- H. Zeng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06651k |
| This journal is © The Royal Society of Chemistry 2014 |