
Asymmetric synthesis towards doxanthrine, a dopamine D1 full agonist†
Rajesh Malhotraa,
Amit Ghoshab,
Swarup Duttab,
Tushar K. Deyab,
Sourav Basu*b and
Saumen Hajra*cd
aDepartment of Chemistry, Guru Jambheshwar University of Science and Technology, Hisar, Haryana 125001, India
bTCG Life Sciences Ltd, Saltlake, Kolkata 700091, India. E-mail: sourav@chembiotek.com; Fax: +91-33-23673058; Tel: +91-33-23673151
cDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: shajra@chem.iitkgp.ernet.in; Fax: +91-3222-282252; Tel: +91-3222-283340
dCentre of Biomedical Research, SGPGIMS Campus, Lucknow 226014, India. E-mail: saumen.hajra@cbmr.res.in
First published on 24th September 2014
Abstract
Asymmetric synthesis of O-methyl doxanthrine is accomplished with high diastereo- and enantioselectivity from β-aryloxyamino acid derived from D-serine.
Dopamine D1 full agonists are considered as potential therapeutic drugs for the treatment of Parkinson's disease.1 Dihydrexidine 1 (Fig. 1) was introduced as the first high affinity bioavailable full dopamine D1 agonist with ten-fold higher affinity for D1-like receptors over D2 like receptors.2 Initially it was examined for the treatment of Parkinson's disease but due to intense hypotension upon intravenous administration the development work on this was halted. Recently an oxygen bioisostere of dihydrexidine, trans-2,3-dihydroxy-6a,7,8,12b-tetrahydro-6H-chromeno[3,4-c]isoquinoline namely doxanthrine 2 (Fig. 1), has been introduced. This showed >200 fold selectivity for D1-like receptors over D2 like receptors.3 More importantly, these compounds are found to exhibit a high level of enantiospecificity in their interaction with the D1 receptor.3b Thus asymmetric synthesis of doxanthrine is in high demand. There are only two reports on synthesis of (±)-doxanthrine and both are by Nichols group,3a,d where non-racemic doxanthrine was obtained by resolution of O-protected doxanthrine via diastereomeric separation of amide of the later and R-(−)-O-methylmandeloyl chloride. First synthesis of (±)-2 was done starting from 4-chromanon using Suzuki coupling, alkene nitration with tetranitromethane and chemoselective reduction of 4-aryl-3-nitro-2H-chromene as the key steps.3a Recently the same group reported an elegant and divergent approach for the racemic synthesis of 2 via 1,4-conjugate addition of ortho-lithiated aryloxazolines to 3-nitro-2H-chromene, which avoids the use of tetranitromethane and halogenated arenes.3d However, there is no report, to the best of our knowledge, on asymmetric synthesis of doxanthrine. Our continuous efforts4,5 toward asymmetric synthesis of dopamine D1 agonists led us to investigate the synthesis of doxanthrine too. Here in, we report first asymmetric synthesis of O-methyl doxanthrine.
 | ||
| Fig. 1 Structural difference in dihydrexidine 1 and doxanthrine 2. | ||
From our earlier study on hexahydrobenzophenanthrene compounds, it reveals that doxanthrine can be synthesized from aminochroman 3, where tethered one-carbon ortho-substitution will be utilized for C-ring formation.4,5 Aminochroman 3 can be obtained from 3-aryloxy-2-aminopropanol 4 via Friedel–Crafts type cyclization. In turn aminoalcohol 4 can be synthesized from β-aryloxyamino acid 5, which can be obtained from D-serine (Scheme 1).
 | ||
| Scheme 1 Retrosynthetic approach of doxanthrine 2. | ||
To begin the synthesis, N-Boc-β-aryloxyamino acid 5 was efficiently synthesized by regio-selective ring opening of sulfamidate carboxylic acid 6 derived from D-serine following our developed method (Scheme 2).6 To avoid the difficulties in acid/Lewis acid mediated Friedel–Crafts cyclization,4d N-Boc protection of aryloxy acid 5 was changed to N-Cbz protected acid 5b, which was transformed to Weinreb amide 7. This amide 7 could be an important and advance precursor for the synthesis of a wide variety of chiral aminochromans and doxanthrine derivatives on reaction with different ArM (M = Li, MgX) followed by Friedel–Crafts cyclization. Our earlier efforts towards syntheses of hexahydrobenzophenanthridine dopamine D1 agonists reveals that aminochroman must have a tethered ortho-substitution phenyl ring.4d,5c We planned for the reaction of the Weinreb amide 7 with ortho-lithiated toluene and also presumed that the tethered ortho-methyl group would be functionalized to construct the C-ring of doxanthrine. For this purpose, ortho-lithiated toluene generated from ortho-bromo toluene was reacted with the Weinreb amide 7 and produced ketone 8 in very good yield. NaBH4 reduction of the ketone 8 afforded excellent yield of amino alcohol 4a as a diastereomeric mixture (dr: 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). We are delighted to report that amino alcohol 4a smoothly underwent Bi(OTf)3 catalyzed Friedel–Crafts reaction and afforded exclusively trans-N-Cbz protected amino chroman 9 (dr: 99:1) in good yield.7 The trans-stereochemistry of aminochroman 9 was determined from the coupling constant of the bibenzylic proton (ArCHAr′) appeared as a doublet at δ 4.03 (J = 8.4 Hz) and on correlation with trans-aminotetralin4a,d and with cis- and trans-tetrahydro chromenoisoquinolin.3a Functionalization of tethered ortho-methyl group and construction of C-ring can lead to doxanthrine moiety. However, our attempt to oxidize the methyl group of chroman 9 to carboxylic acid using KMnO4 and SeO2 was failed to give any desired acid and always led to a complex mixture of unidentified compounds. Peroxide as well as AIBN mediated benzylic bromination with NBS was also unsuccessful, mostly provided (mono- and di-) bromination of electron-rich trioxyarene (ring A).
:1). We are delighted to report that amino alcohol 4a smoothly underwent Bi(OTf)3 catalyzed Friedel–Crafts reaction and afforded exclusively trans-N-Cbz protected amino chroman 9 (dr: 99:1) in good yield.7 The trans-stereochemistry of aminochroman 9 was determined from the coupling constant of the bibenzylic proton (ArCHAr′) appeared as a doublet at δ 4.03 (J = 8.4 Hz) and on correlation with trans-aminotetralin4a,d and with cis- and trans-tetrahydro chromenoisoquinolin.3a Functionalization of tethered ortho-methyl group and construction of C-ring can lead to doxanthrine moiety. However, our attempt to oxidize the methyl group of chroman 9 to carboxylic acid using KMnO4 and SeO2 was failed to give any desired acid and always led to a complex mixture of unidentified compounds. Peroxide as well as AIBN mediated benzylic bromination with NBS was also unsuccessful, mostly provided (mono- and di-) bromination of electron-rich trioxyarene (ring A).
 | ||
| Scheme 2 Synthesis of aminochroman 9. Reagents and conditions: (a) 3,4-(MeO)2C6H3OH (0.8 eq.), NaH (3.2 equiv.), THF, −15 to 0 °C; (b) (i) TFA, CH2Cl2, 0 °C; (ii) Cbz-succinimide (1.0 equiv.), Et3N (4.0 equiv.)), THF–H2O (3:1), r.t., 12 h; (c) NMe(OMe)·HCl (1.5 equiv.), ClCO2Bui (1.2 equiv.), NMM (2.2 equiv.), CH2Cl2, −15 °C to rt, 5 h; (d) 2-BrC6H4Me (3.2 equiv.), n-BuLi (3.0 equiv.), THF, −78 °C; (e) NaBH4 (1.0 equiv.), EtOH; (f) Bi(OTf)3 (0.3 eq.), MeNO2, 45 °C, 1 h. | ||
To avoid the difficulties in functionalization of ortho-methyl group, O-benzyl ortho-lithiated benzyl alcohol generated from 1-((benzyloxy)methyl)-2-bromobenzene 11 and n-BuLi was reacted with the Weinreb amid 7 and provided moderate yield of ketone 12 (Scheme 3). Reduction of ketone 12 with NaBH4 gave diastereomeric mixture of alcohol 13 (dr: 3:1). Unlike alcohol 4, Bi(OTf)3 catalyzed Friedel–Crafts cyclization of alcohol 13 showed <10% of cyclised product chroman 14 in LC-MS analysis. A variety of acids and Lewis acids such as TFA, MeSO3H, PTSA, PPA, PhSO3H, FeCl3, Cu(OTf)2, CuCl2 etc. were tested for Friedel–Crafts cyclization of alcohol 13. Most of the reactions showed a complex mixture of unidentified compounds, few cases did show desired mass in LC-MS, but as a minor product (5–10%). Poor yield of the Friedel–Crafts cyclization might be due to ortho-setric effect of tethered –CH2OBn unit and its side reactions, indicated by the presence of mass (m/z) of debenzylated 13 in LC-MS of the above reaction mixtures.
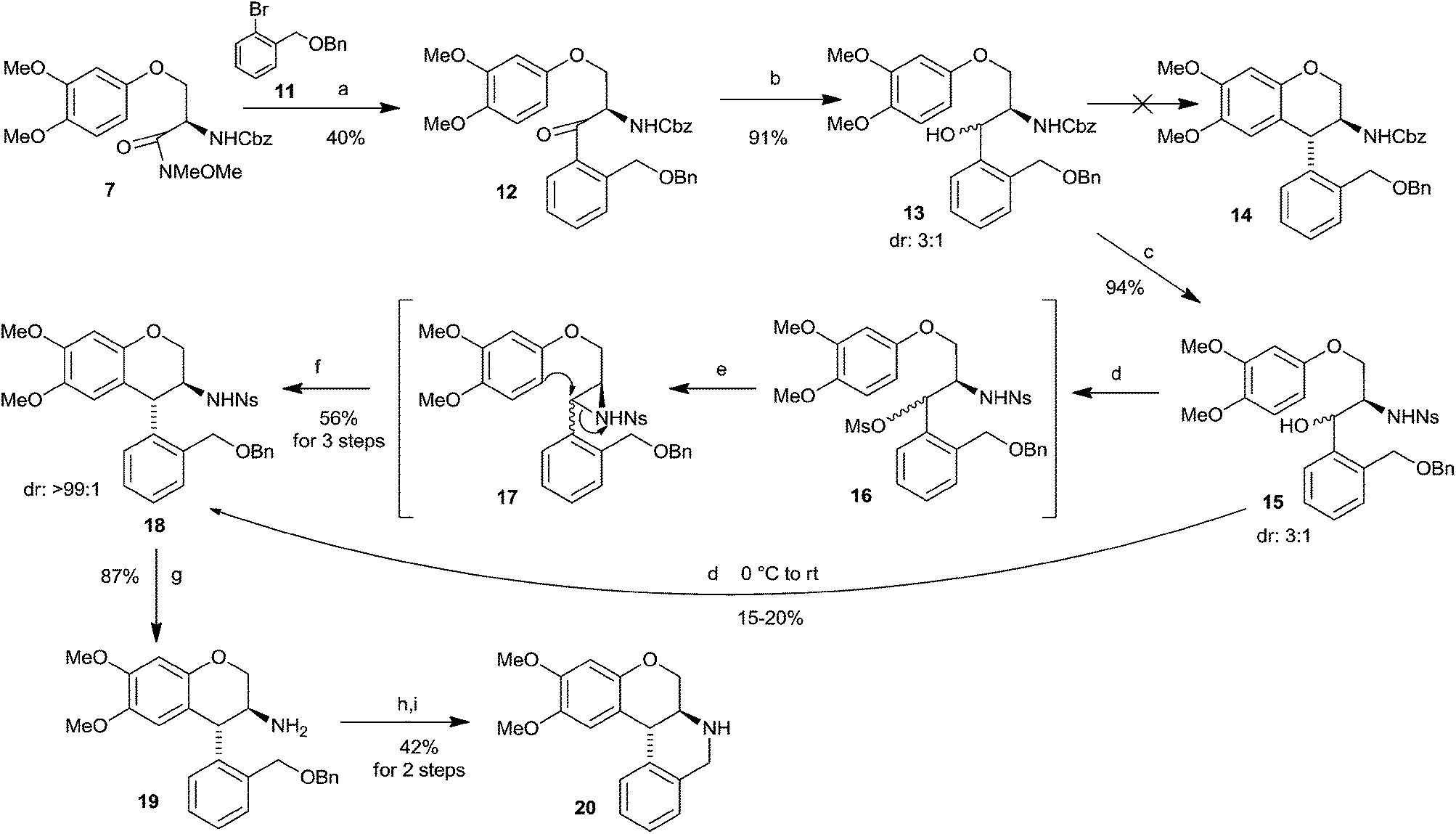 | ||
| Scheme 3 Asymmetric synthesis of O-methyl doxanthrine. Reaction conditions and reagents: (a) ArBr 11 (3.2 equiv.), n-BuLi (3.0 equiv.), THF, −78 °C; (b) NaBH4 (1.0 equiv.), EtOH; (c) (i) H2 (1 atm), 10% Pd/C, MeOH, rt, 1 h; (ii) NsCl (1.1 equiv.), Et3N (3.0 equiv.), CH2Cl2, 0 °C to rt; (d) MsCl (1.7 equiv.), Et3N (3.0 equiv.), CH2Cl2 (e) NaH (1.0 equiv.), THF, 50 °C, 4 h; (f) Cu(OTf)2, ClCH2CH2Cl, 70 °C, 2 h, 56%; (g) 4-MeOC6H4SH (3.0 equiv.), K2CO3 (5.0 equiv.), CH3CN–DMSO (49:1), rt, 5 h; (h) H2 (1 atm), 10% Pd/C, AcOH, rt, 8 h; (i) (i) HCl, 1,4-dioxane, reflux, 2 h; (ii) K2CO3 (40.0 equiv.), 1,4-dioxane, 80 °C, 1 h. | ||
Stereo- and regioselective aminoarylation towards synthesis of aminochromans could also be an alternative strategy.5c As a requirement, the N-Cbz amino alcohol 13 transformed to N-nosyl amino alcohol 15 via Pd/C hydrogenation and nosyl protection. Alcohol 15 on reaction with MsCl and Et3N produced O-mesylated compound. Mesylate 16 was expected to undergo Friedel–Crafts cyclization providing desired aminochroman. When the mesylation reaction was continued at 0 °C and at rt, we did observe the formation of 15–20% of aminochroman 18 along with a mixture of unidentified compounds in LC-MS. However, prolonging the reaction at 0 °C and also at rt did not improve the yield of cyclised product rather it led to a complex mixture of unidentified compounds. This might be due to presence of three oxybenzyl units. It is thought that the mesylate 16 can be transformed to aziridine 17, which would undergo aminoarylation (Friedel–Crafts cyclization) under mild condition. As the alcohol 15 is a mixture of diastereomers (dr: 3:1), the intermediate aziridine 17 is expected to be a mixture of diastereomers with same ratio (dr: 3:1). It is well established in our previous study that trans-aziridine undergoes faster aminoarylation (Friedel–Crafts cyclization) via SN2 type mechanism, where as cis-isomer is not reactive to SN2 ring-opening and undergoes an SN1 ring-opening mechanism followed by intramolecular Friedel–Crafts reaction to give the more stable trans-product.8 Thus the cis-isomer needs little harsh conditions compare to trans-aziridine, but both provide exclusively trans-cyclized product. Accordingly the mesylate of 16, without any purification, was treated with NaH and heated at 50 °C. We are delighted to report that it gave directly aminochroman 18 along with minor amount of an intermediate azidirine 17 as a non-separable mixture. The crude mixture in ClCH2CH2Cl was treated with catalytic amount of Cu(OTf)2 and it fully transformed to trans-amino chroman 18 with excellent diastereoselectivity (dr: >99:1) in 56% of yield over three steps from amino alcohol 15. trans-Stereochemistry of aminochroman 18 was assigned from the coupling constant of ArCHAr′ appeared as a doublet at δ 3.94 with coupling constant of 10.4 Hz, and comparing with that of trans-aminotetralin4a,d and with cis- and trans-tetrahydro chromenoisoquinolin.3a Thus confirms the absolute stereochemistry of compound 18 as 3S, 4R. N-Nosyl group of chroman 18 was removed on treatment with 4-methoxythiophenol and K2CO3 and afforded aminochroman 19 in 87% of yield. For the construction of C-ring, debenzylation of compound 19 by hydrogenation in the presence of Pd/C produced amino alcohol. Without any purification it was heated with HCl in dioxane followed by treatment with K2CO3 accomplished the synthesis of O-methyl doxanthrine 20 as an off-white solid in 42% of yield over two steps. Bibenzylic proton (ArCHAr′) of compound 20 appeared as a doublet at δ 3.94 (J = 10.9 Hz), which was compared with literature data, where the ArCHAr′ of methylenedioxy doxanthrine showed at δ 4.03 (d, J = 11.4) and the corresponding cis-isomer at δ 3.89 (d, J = 6.0 Hz).3a Thus it determines the trans-stereochemistry of O-methyl doxanthrine and in turn confirms the absolute stereochemistry of compound 20 as 6aS,12bR. Optical rotation of the O-methyl doxanthrine 20 was found to be +76.2 (c 1.0, CH2Cl2). Demethylation of compound 20 would accomplish the synthesis of doxanthrine 2. O-Methyl doxanthrine 20 was treated with BBr3 under ice-cold conditions. It showed the formation of desired demethylated product along with a mixture of uncharacterized compounds, but the doxanthrine 2 could not be isolated as a pure form. HBr, HI and AlCl3 instead of BBr3 gave a complex mixture of identified compounds and there was no reaction with BCl3 and Py·HCl under different reaction conditions.
In conclusion, we have achieved first asymmetric synthesis of O-methyl doxanthrine from β-aryloxy amino acid derived from D-serine using metaloarene addition to Weinreb amide and Friedel–Crafts cyclization as the key steps. Accomplishment to the total synthesis of doxanthrine via demethylation was not successful. It may need milder condition for the demethylation or instead of methoxy group some other stable, but easily removable phenolic protection group. The developed protocol provides an easy avenue for the asymmetric synthesis of a variety of aminochromans and tetrahydrochromeno-isoquinolines by varying the phenols in regioselective opening of serine sulfamidate and in metaloarenes addition to Weinreb amide.
Acknowledgements
We thank DBT (BIPP), New Delhi (sanction no. 102/IFD/SAN/1191/2009-2010) for providing financial support.Notes and references
- (a) W. J. Giardina and M. Williams, CNS Drug Rev., 2001, 7, 305 CrossRef CAS PubMed; (b) S. Peter, I. Ruben and K. Bjorn, CNS Drug Rev., 2004, 10, 230 Search PubMed; (c) M. Malo, L. Brive, K. Luthman and P. Svenssonm, Chem. Med. Chem., 2012, 7, 483 CrossRef CAS PubMed; (d) J. Zhang, B. Xiong, X. Zhen and A. Zhang, Med. Res. Rev., 2009, 29, 272 CrossRef CAS PubMed.
- (a) W. K. Brewster, D. E. Nichols, R. M. Riggs, D. M. Mottola, T. W. Lpvenberg, M. H. Lewis and R. B. Mailman, J. Med. Chem., 1990, 33, 1756 CrossRef CAS PubMed; (b) J. R. Taylor, M. S. Lawrence, D. E. Redmont, J. D. Elsworth, R. H. Roth, D. E. Nochols and R. B. Mailman, Eur. J. Pharmacol., 1991, 199, 389 CrossRef CAS PubMed; (c) M. P. Seiler, A. Hagenbach, H.-J. Wüthrich and R. Marksteun, J. Med. Chem., 1991, 34, 303 CrossRef CAS PubMed; (d) T. A. Knoerzer, D. E. Nichols, W. K. Brewster, V. J. Watts, D. Mottola and R. B. Mailman, J. Med. Chem., 1994, 37, 2453 CrossRef CAS PubMed; (e) M. R. Michaelides, Y. Hong Jr, S. DiDomenico, K. E. Asin, D. R. Britton, C. W. Lin, M. Williams and K. Shiosaki, J. Med. Chem., 1995, 38, 3445 CrossRef CAS PubMed; (f) J. G. Berger, W. K. Chang, J. W. Clader, D. Hou, R. E. Chipkin and A. T. McPhail, J. Med. Chem., 1989, 32, 1913 CrossRef CAS PubMed.
- (a) J. P. Cueva, G. Giorgioni, R. A. Grubbs, B. R. Chemel, V. J. Watts and D. E. Nichols, J. Med. Chem., 2006, 49, 6848 CrossRef CAS PubMed; (b) J. A. Przybyla, J. P. Cueva, B. R. Chemel, K. J. Hsu, D. J. Riese, J. D. McCorvy, J. A. Chester, D. E. Nichols and V. J. Watts, Eur. Neuropsychopharmacol., 2009, 19, 138 CrossRef CAS PubMed; (c) J. D. McCorvey, V. J. Watts and D. E. Nichols, Psychopharmacology, 2012, 222, 81 CrossRef PubMed; (d) J. P. Cueva, B. R. Chemel, J. I. Juncosa, M. A. Lill, V. J. Watts and D. E. Nichols, Eur. J. Med. Chem., 2012, 48, 97 CrossRef CAS PubMed.
- (a) R. Malhotra, A. Ghosh, R. Ghosh, S. Chakrabarti, S. Dutta, T. K. Dey, S. Roy, S. Basu and S. Hajra, Tetrahedron: Asymmetry, 2011, 22, 1522 CrossRef CAS ; Corrigendum: R. Malhotra, A. Ghosh, R. Ghosh, S. Chakrabarti, S. Dutta, T. K. Dey, S. Dutta, S. Roy, S. Basu and S. Hajra, Tetrahedron: Asymmetry, 2013, 24, 341 Search PubMed; (b) R. Malhotra, S. Chakrabarti, A. Ghosh, R. Ghosh, S. Dutta, T. K. Dey, S. Roy, S. Basu and S. Hajra, Tetrahedron: Asymmetry, 2013, 24, 278 CrossRef CAS; (c) S. Hajra, R. Ghosh, S. Chakrabarti, A. Ghosh, S. Dutta, T. K. Dey, R. Malhotra, S. Asijaa, S. Roy, S. Dutta and S. Basu, Adv. Synth. Catal., 2012, 354, 2433 CrossRef CAS; (d) R. Malhotra, S. Chakrabarti, T. K. Dey, S. Dutta, K. B. Alapatti, S. Dutta, S. Roy, S. Basu and S. Hajra, Tetrahedron, 2013, 69, 8994 CrossRef CAS.
- (a) S. Hajra and S. Bar, Chem. Commun., 2011, 47, 3981 RSC; (b) S. Hajra and S. Bar, Tetrahedron: Asymmetry, 2011, 22, 775 CrossRef CAS ; Corrigendum: S. Hajra and S. Bar, Tetrahedron: Asymmetry, 2013, 24, 340 Search PubMed; (c) S. Hajra and D. Sinha, J. Org. Chem., 2011, 76, 7334 CrossRef CAS PubMed.
- R. Malhotra, T. K. Dey, S. Dutta, S. Basu and S. Hajra, Org. Biomol. Chem., 2014, 12, 6507 CAS.
- D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557 CrossRef CAS PubMed.
- S. Hajra, B. Maji and D. Mal, Adv. Synth. Catal., 2009, 351, 859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and LC-MS for all the compounds 4–9, 12, 13, 15, 18–20. See DOI: 10.1039/c4ra06617k |
| This journal is © The Royal Society of Chemistry 2014 |