
The influence of the leaving iodine atom on phyllosilicate syntheses and useful application in toxic metal removal with favorable energetic effects†
Abstract
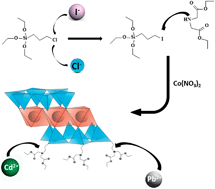
Novel nanostructured 2 : 1 hybrid cobalt and nickel phyllosilicates containing attached diethyl iminodiacetate moieties within the interlamellar spaces were synthesized using a mild non-hydrothermal sol–gel methodology. The organofunctionalization of these hybrids was achieved using silylating agents formed by the reaction of 3-chloropropyltriethoxysilane or 3-iodopropyltrietoxysilane with diethyl iminodiacetate, whereas 3-iodopropyltriethoxysilane was synthesized from the reaction of 3-chloropropyltriethoxysilane with NaI. The degrees of functionalization of the new materials are associated with the nucleophilic displacement of the halide atom by the nitrogen basic center of diethyl iminodiacetate during the silylating agents' syntheses. The incorporation of an iodine atom favored the introduction of bulky basic moieties in the pendant chain. For example, the degree of functionalization for the nickel phyllosilicate functionalized from 3-iodopropyltriethoxysilane, (1.83 ± 0.01) mmol g−1, was higher than that of the nickel phyllosilicate functionalized from 3-chloropropyltriethoxysilane, (0.59 ± 0.02) mmol g−1. Infrared spectroscopy combined with 13C and 29Si NMR spectroscopy confirmed the attachment of organic moieties covalently bonded to the silicon sheet network, and the lamellar 2 : 1 trioctahedral phyllosilicate structures were confirmed by XRD. Further characterization was provided by thermogravimetry, SEM and TEM, which exhibited thermally stable hybrids presenting well-formed particles with a homogeneous distribution of cobalt, nickel, and nitrogen. The attached basic centers have the ability to sorb lead and cadmium from aqueous solution. The sorption data fit the Langmuir model and indicated maximum lead sorption values of (2.11 ± 0.16) mmol g−1 for the cobalt phyllosilicate functionalized from 3-iodopropyltriethoxysilane and (1.99 ± 0.11) mmol g−1 for the hybrid prepared from 3-chloropropyltriethoxysilane, for example, reflecting the higher degree of functionalization of the former. All of the other hybrids exhibited the same tendency, even for cadmium sorption. The thermodynamic data obtained from calorimetric titration revealed the spontaneity of these chelating interactive processes, which are enthalpically and entropically favorable for the proposed cation-basic center interactions at the solid–liquid interface.
 Please wait while we load your content...
Please wait while we load your content...

