
The construction of a fluorescent nano-probe and its application in detecting transgenic Bt rice TT51-1
Abstract
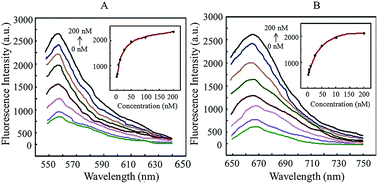
In this study, we used 13 nm gold nanoparticles (AuNPs) as the quenching group and Cy3/Cy5 fluorescent dye as the reporting group to develop a non-PCR method to detect transgenic rice TT51-1. The results showed that it had high sensitivity and specificity, and was time-efficient, inexpensive and simple to use.
 Please wait while we load your content...
Please wait while we load your content...

