
Greener and expeditious one-pot synthesis of dihydropyrimidinone derivatives using non-commercial β-ketoesters via the Biginelli reaction†
Abstract
An expeditious synthesis of novel 3,4-dihydropyrimidin-2(1H)-one derivatives has been developed using a multi-component reaction involving the in situ generation of non-commercial β-ketoesters via transesterification of tert-butyl-β-ketoester with corresponding alcohol followed by the Biginelli reaction with arylaldehyde and urea in one-pot at 110 °C under green conditions.
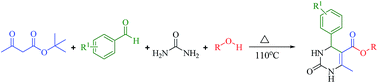
 Please wait while we load your content...
Please wait while we load your content...

