
Effect of melting temperature on interfacial interaction and mechanical properties of polypropylene (PP) fiber reinforced olefin block copolymers (OBCs)
Abstract
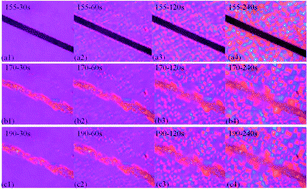
PP fiber reinforced olefin block copolymers (OBCs) were manufactured in this research. Chopped PP fibers were first compounded with OBCs at a fixed ratio in a Haake mixer at 140 °C (lower than the melting point of PP fiber). The blends were then processed at three different temperatures, below (155 °C), around (170 °C) and above (190 °C) the melting temperature of the fibers. The three specimens displayed completely different mechanical properties according to the following sequence: 170 °C > 190 °C > 155 °C. To obtain an insight into the phenomenon, the structure–property relationships were fully investigated. It was found that when the blends were processed at 155 °C, the fibers retained a high strength and modulus but lost the interaction with the matrix, displaying poor mechanical properties. At a higher temperature of 190 °C, the fiber showed a strong interaction with OBCs but the reinforcing effect was unsatisfactory because the melted fibers lost the original modulus. When the blends were injection molded at 170 °C, the fibers were in the partially melted state, showing strong interaction with the matrix and relatively high strength and modulus. Therefore, the composites exhibited the mechanical performance. This article proves that the fiber strength and interfacial bonding are the two factors that determine the final properties in fiber reinforced composites. Moreover, it is suggested that the interfacial interaction could be improved by controlling the melting state instead of complex surface treatment of the fibers.
 Please wait while we load your content...
Please wait while we load your content...

