
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How much do coulombic interactions stabilize a mesophase? Ion pair and non-ionic binary isosteric derivatives of monocarbaborates and carboranes†
Aleksandra
Jankowiak
a,
Ajan
Sivaramamoorthy
a,
Damian
Pociecha
b and
Piotr
Kaszyński
*ac
aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. E-mail: piotr.kaszynski@vanderbilt.edu; Tel: +1-615-322-3458
bDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cFaculty of Chemistry, University of Łódź, Tamka 12, 91403 Łódź, Poland
First published on 15th October 2014
Abstract
Replacement of the B− atom in the monocarbaborate anion, 1[10] or 1[12], and the N+ atom in the pyridinium cation [Pyr] of a liquid crystalline ion pair with C atoms leads to an isoelectronic and isosteric non-ionic binary liquid crystalline mixture of carborane (2[10] or 2[12]) and benzene ([Ph]) derivatives lacking coulombic interactions. A comparison of mesogenic properties of ion pairs, 1[10]c–[Pyr]c and 1[12]c–[Pyr]c, with their analogous non-ionic mixtures, 2[10]c–[Ph]c and 2[12]c–[Ph]c, shows a 181 K higher clearing temperature, Tc, for the ion pair. This corresponds to a DFT-calculated difference in association energy ΔΔHa = 24.5 kcal mol−1 in a typical dielectric medium (ε = 2.5). Pure compounds and binary mixtures were characterized using thermal, optical, and XRD methods.
Introduction
Ionic liquid crystals (ILC) are capable of anisotropic ion transport,1–4 and for this reason they are becoming increasingly attractive electrolytes for ion batteries,5 photovoltaic6–8 and other applications.9,10 In contrast to non-ionic mesogens, typical ILC are binary systems, comprising of a cation and an anion. Consequently, mesogenic properties of ILC are controlled not only by the anisometry of the two components, but also by electrostatic interactions.11 While the first factor is well understood,12 the effect of coulombic interactions on phase stability is poorly characterized and has only recently become the subject of theoretical considerations.13,14 Despite the large number of examples of ILC,10 there exists no single report of a molecular system that permits experimental evaluation of the coulombic component of the phase stability in the absence of other factors. We now report such a molecular system and for the first time assess the effect of coulombic interactions, as the only variable, on mesophase stability in ILC.During the past several years, we have developed new ILC materials in which anisometric anions derived from monocarbaborates, [closo-1-CB9H10]− ([10]) and [closo-1-CB11H12]− ([12]), are the driving force to induce mesogenic behaviour.15–18 Due to the high delocalization of the charge and the resulting weak nucleophilicity of the boron cluster anions,19 the ion interactions in such ILC, e.g.1a–[Pyr]a (Fig. 1),16 are non-specific and mainly electrostatic. Such ion pairs typically form SmA and soft crystalline phases, although some exhibit a nematic phase, e.g. azo derivative 1[12]b–[Pyr]a (Fig. 1).16
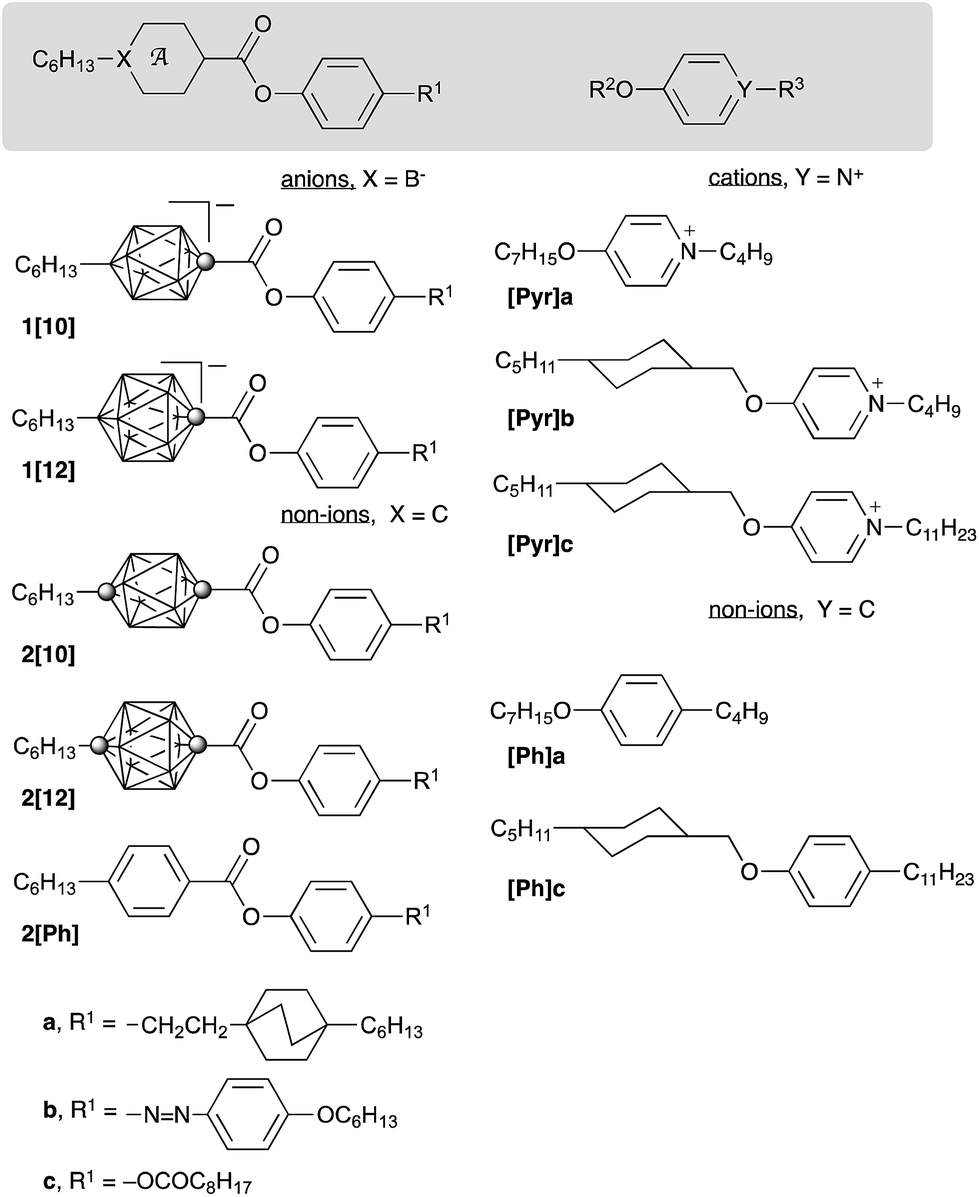 | ||
| Fig. 1 The structure of ions (1 and [Pyr]) and non-ions (2 and [Ph]). In the cage structures, each vertex represents a BH fragment, and the sphere is a carbon atom. | ||
Replacement of the B− (12) atom in anion 1[12]b and the N+ atom in cation [Pyr]a with C atoms leads to a non-ionic pair consisting of carborane 2[12]b and benzene derivative [Ph]a. An equimolar mixture of 2[12]b and [Ph]a represents an isosteric, non-coulombic analogue of the mesogenic ion pair 1[12]b–[Pyr]a.
Here, we report the synthesis and detailed thermal, optical and structural (XRD) analysis of ion pairs 1[10]–[Pyr] and 1[12]–[Pyr] as well as their electrically neutral analogues, 2 and [Ph]. We emphasize the comparison of the mesogenic behaviour of ion pairs 1–[Pyr] with the isosteric, non-ionic equimolar mixtures 2–[Ph]. The experimental results are supported with DFT modelling of the association of ions and non-ionic analogues in a dielectric medium.
Results and discussion
Synthesis
Esters 1c and 2c were obtained by reaction of phenol 3c (ref. 15) with appropriate acid chlorides which were prepared from carboxylic acids 4[10],154[12],165[10] (ref. 20) and 5[12] (Method A, Scheme 1). Without isolation, ion pairs 1c–[NEt4], obtained from acid 4[10]–[NEt4] (ref. 15) and 4[12]–[NEt4],16 were converted to 1c–[Pyr] by cation exchange with [Pyr]Br as described before for esters 1a–[Pyr]a and 1b–[Pyr]a.15,16 Esters 2a and 2b were prepared directly from phenols 3a (ref. 16) and 3b,16 respectively, and carboxylic acid 5 in the presence of DCC (Method B).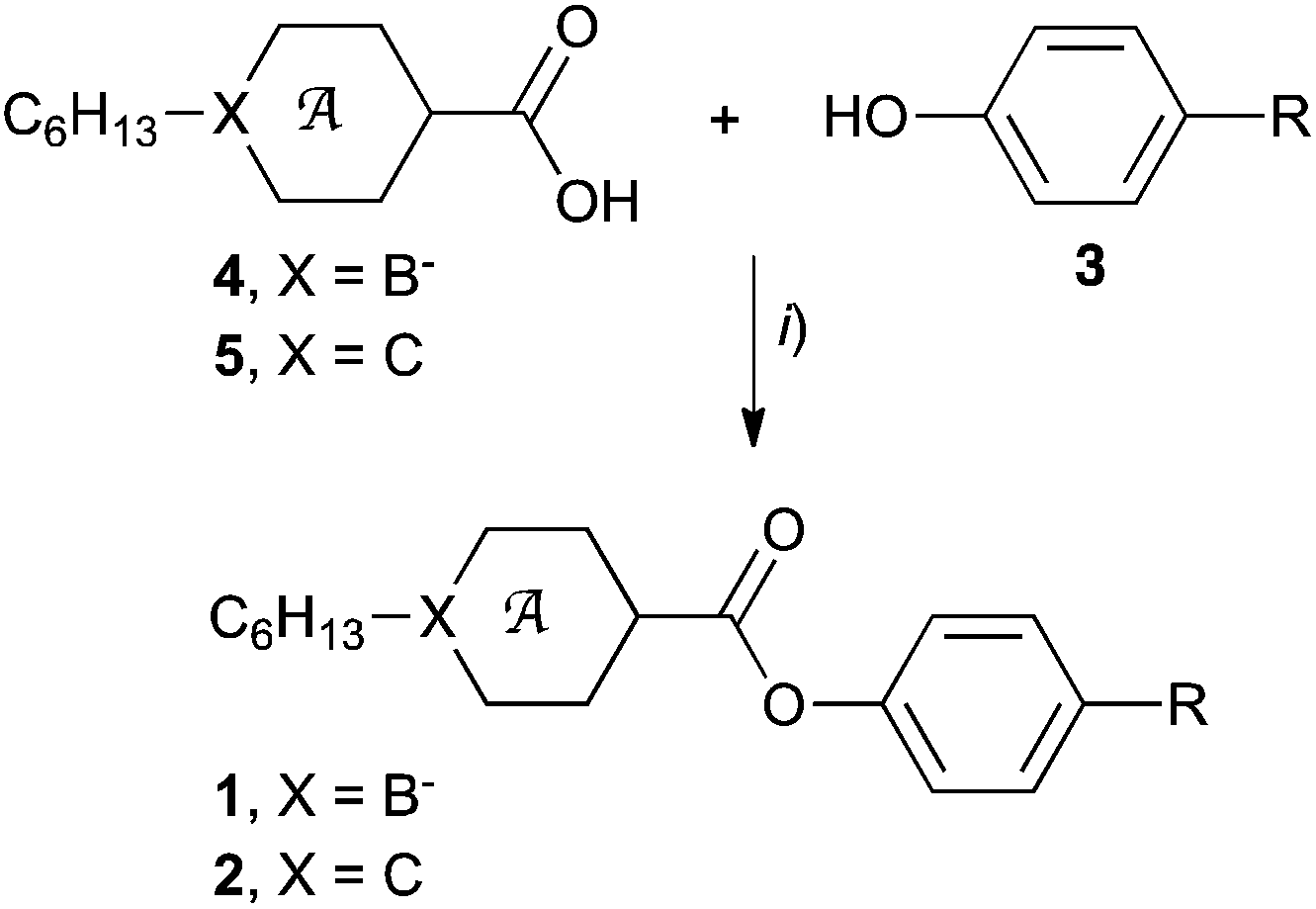 | ||
| Scheme 1 Synthesis of esters 1 and 2. Reagents and conditions: (i) Method A: (1) (COCl)2, cat. DMF, CH2Cl2; (2) Et3N, CH2Cl2 or Method B: DCC, DMAP, CH2Cl2, rt, 12 h. | ||
12-Hexyl-p-carborane-1-carboxylic acid (5[12]) was obtained by alkylation followed by carboxylation of p-carborane (Scheme 2) as described before for the pentyl analogue.21
 | ||
| Scheme 2 Synthesis of carboxylic acid 5[12]. Reagents and conditions: (i) (1) BuLi 1 eq, THF −80 °C → rt → −80 °C; (2) C6H13I 1 eq, 0 °C; (3) BuLi 1 eq, −80 °C → 0 °C; (4) CO2, (5) HCl. | ||
Pyridinium bromides [Pyr]b–Br and [Pyr]c–Br were prepared by N-alkylation of 4-(trans-4-pentylcyclohexylmethyloxy)-pyridine (6) with appropriate alkyl bromide in MeCN following the procedure described for [Pyr]a–Br (Scheme 3).15 The substituted pyridine 6 was obtained from 4-chloropyridine and trans-4-pentylcyclohexylmethanol22 (7) in the presence of NaH in DMSO following a general method.23
 | ||
| Scheme 3 Synthesis of pyridinium salts [Pyr]–Br. Reagents and conditions: (i) NaH, DMSO, rt; (ii) CnH2n+1Br, MeCN, reflux. | ||
The cyclohexylmethyl phenyl ethers [Ph] were obtained by alkylation of appropriate 4-alkylphenol (8[n]) with heptyl tosylate (for [Ph]a) or trans-4-pentylcyclohexylmethyl tosylate229 for [Ph]b and [Ph]c (Scheme 4). Phenol 8[11] was obtained by alkylation of 4-bromophenol with C11H23MgBr following a general procedure.24
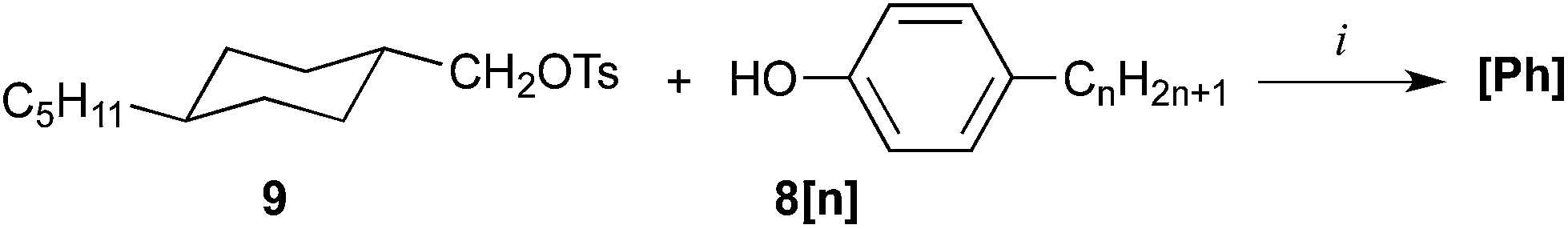 | ||
| Scheme 4 Synthesis of phenyl ethers [Ph]. Reagents and conditions: (i) K2CO3, MeCN, cat Aliquat, reflux. | ||
Thermal analysis
Transition temperatures and enthalpies of the newly prepared compounds were determined by differential scanning calorimetry (DSC) and results are shown in Tables 1 and 2.|
|
||||||
|---|---|---|---|---|---|---|
| R1 | A | Cr | N | I | ||
| a Cr-crystal, N-nematic, I-isotropic. Temperatures obtained on heating at 5 K min−1. Enthalpies are listed in the ESI. b Cr1 104 Cr2. c Ref. 26. d Ref. 17. e Cr1 41 Cr2. | ||||||

|
[10] | ˙ | 114b | ˙ | 175 | ˙ |
| [12] | ˙ | 145 | ˙ | 187 | ˙ | |
| [Ph] c | ˙ | 89.5 | ˙ | 200 | ˙ | |

|
[10] | ˙ | 67 | ˙ | 197 | ˙ |
| [12] | ˙ | 85 | ˙ | 205 | ˙ | |
| [Ph] d | ˙ | 91 | ˙ | 206 | ˙ | |
|
[10] | ˙ | 44e | (˙ | 36) | ˙ |
| [12] | ˙ | 46 | (˙ | 45) | ˙ | |
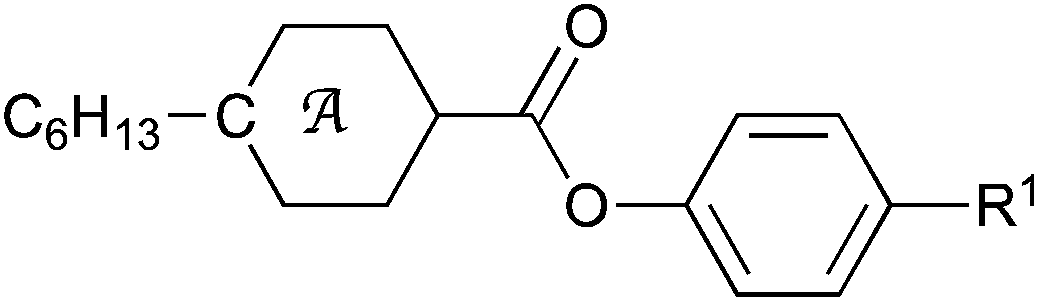
| X | Y | Cr | E | SmA | N | I | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Cr-crystal, SmA-smectic A, E-soft crystalline phase, N-nematic, I-isotropic. Temperatures obtained on heating at 5 K min−1. Enthalpies are listed in the ESI. For definition of substituents R see Fig. 1. b Cr1 85 Cr2. c Ref. 16. d Cr1 64 Cr2 101 × 107. | |||||||||||
| 1[10]c–[Pyr]c | B− | N+ | ˙ | 126 | ˙ | 163 | ˙ | 207 | ˙ | ||
| 2[10]c–[Ph]c | C | C | ˙ | 27 | (˙ | 26) | ˙ | ||||
| 1[12]c–[Pyr]c | B− | N+ | ˙ | 148b | ˙ | 200 | ˙ | 210 | ˙ | ||
| 2[12]c–[Ph]c | C | C | ˙ | 30 | (˙ | 29) | ˙ | ||||
| 1[10]c–[Pyr]a c | B− | N+ | ˙ | 107d | ˙ | ||||||
| 1[12]c–[Pyr]a c | B− | N+ | ˙ | 117 | ˙ | ||||||
| 1[12]c–[Pyr]b | B− | N+ | ˙ | 132 | ˙ | 172 | ˙ | ||||
The phenyl ether [Ph]c is a new member of a known homologous series27 and it exhibits a mesophase identified by XRD analysis as a soft crystalline B phase (Cr 32 SmBcryst 42 I, Fig. 2a).
 | ||
| Fig. 2 The optical texture of (a) B phase in [Ph]c at 29 °C and (b) nematic phase at 25 °C in 2[12]c–[Ph]c obtained on cooling from the isotropic phase. | ||
DSC, POM, and XRD28 analyses revealed that pyridinium bromide [Pyr]c–Br exhibits a SmA phase and another more ordered unidentified phase. Unfortunately, the salt decomposes at about 165 °C. The shorter analogue, [Pyr]b–Br, is not mesogenic.
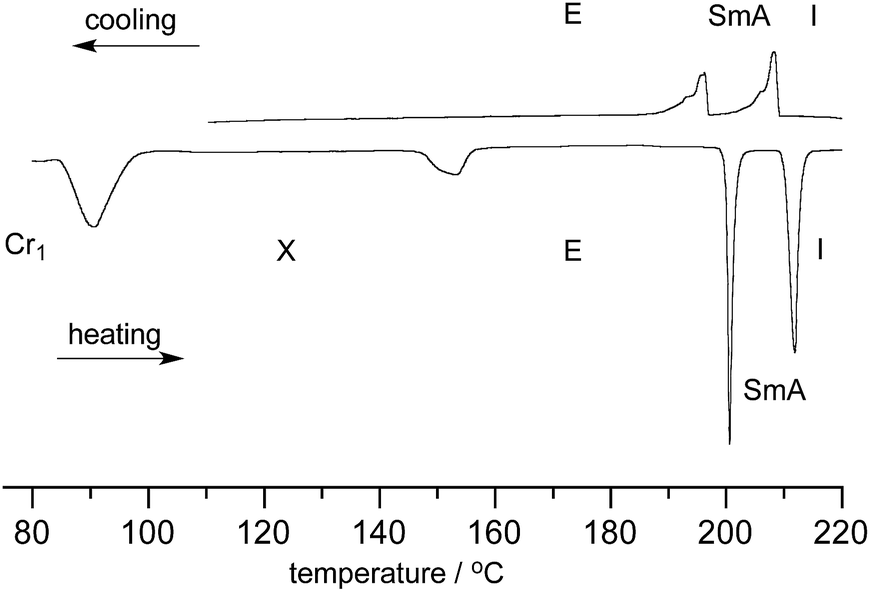 | ||
| Fig. 3 DSC trace of 1[12]c–[Pyr]c. The heating and cooling rates are 5 K min−1. | ||
 | ||
| Fig. 4 The optical textures of (a) SmA phase (205 °C) and (b) E phase (175 °C) obtained for 1[12]c–[Pyr]c on cooling from the isotropic phase in the same region of the sample. | ||
These findings are in agreement with our general observations that a total of 4 rings in the ion structures are necessary for induction of mesogenic behavior in ILC of the general structure 1.16,18 For instance, while 1c–[Pyr]a, a 3-ring ionic compound, does not exhibit mesogenic behaviour, 4-ring ion pairs 1a–[Pyr]a and 1b–[Pyr]a do exhibit enantiotropic SmA and N phases.16
For comparison purposes, equimolar mixtures of non-ionic analogues of 1–[Pyr] were investigated. Initial studies of 2a–[Ph]a and 2b–[Ph]a demonstrated phase separation upon cooling from the isotropic phase. It was considered that the significant difference in the molecular length and mesogenic behaviour of the two components leads to their incompatibility. Therefore, carboranes 2c and phenyl ether [Ph]c were considered, since the total length of the alkyl chains in each compound is 16 atoms.
POM analysis of equimolar mixtures 2[10]c–[Ph]c and 2[12]c–[Ph]c demonstrated ideal miscibility, and the formation of a monotropic nematic phase with clearing temperatures, TNI, of 26 °C and 29 °C, respectively (Table 2). A comparison of these transition temperatures with those obtained for the analogous ion pair 1c–[Pyr]c shows a difference in the clearing temperature of 181 K. Since the ion-pair and the non-ionic binary mixture differ only by a molecular charge, the increase in the clearing temperature can be attributed solely to the strong coulombic interactions present in the ion pair and absent in the non-ionic binary mixture.
XRD data
The formation of the SmA phase by ion pairs 1[10]c–[Pyr]c and 1[12]c–[Pyr]c was confirmed by XRD measurements. Diffractograms obtained for the high temperature mesophase in each material showed a series of sharp commensurate reflections consistent with a lamellar structure (Fig. 5). Considering the calculated molecular length28 of about 33.8 Å for both anions 1[10]c and 1[12]c (including van der Waals radius for the terminal H atoms) the measured layer spacing of about 27 Å indicates approximately 20% of interdigitation and/or imperfect orientational order of molecular long axes and molecular folding in both ion-pairs. | ||
| Fig. 5 Integrated XRD patterns taken in SmA phase of 1[10]c–[Pyr]c (T = 175 °C, upper red curve) and 1[12]c–[Pyr]c (T = 205 °C, lower black curve). Upper curve is vertically shifted for clarity of presentation. | ||
The wide-angle region of the diffractograms shows an unsymmetric broad halo, which can be deconvoluted into two signals with the maxima about 4.7 Å and 5.1 Å for 1[10]c–[Pyr]c, and 4.8 and 5.3 for 1[12]c–[Pyr]c. The diffused signals correlate with the mean distance between the alkyl chains (former) and the separation between boron clusters (latter).
XRD patterns obtained for oriented samples of 1[10]c–[Pyr]c and 1[12]c–[Pyr]c in the phase formed below the SmA phase could be indexed as an E phase (Table 3 and Fig. 6). However, signals from the full size of the in-plane unit cell were also visible,28 which are typically forbidden for E phase. Signals (100) and (010) are observed due to the binary nature of the ionic materials. This causes the molecule in the center of the unit cell to be different than those in the corners resulting in a unit cell that is not centered. There is also a slight difference between patterns for 1[10]c–[Pyr]c and 1[12]c–[Pyr]c. The pattern for the 12-vertex analogue indicates a regular ABA type stacking of the smectic layers, which leads to a doubling of the unit cell dimension along layer normal (thus doubling of the Miller index l, see Fig 6b). The structure of the E phase formed by ion pair 1[12]c–[Pyr]b is similar to that of 1[10]c–[Pyr]c.
| Temperature/°C | a/Å | b/Å | c/Å | |
|---|---|---|---|---|
| 1[10]c–[Pyr]c | 150 | 8.99 | 6.12 | 29.84 |
| 1[12]c–[Pyr]b | 150 | 9.11 | 6.44 | 25.26 |
| 1[12]c–[Pyr]c | 180 | 9.16 | 6.50 | 57.42 |
 | ||
| Fig. 6 2D XRD patterns for E phase of (a) 1[10]c–[Pyr]c at 150 °C and (b) 1[12]c–[Pyr]c at 180 °C. Note the presence of equatorial signals (100) and (010) in (a) and their absence in (b) indicating regular ABA layer stacking for 1[12]c–[Pyr]c. | ||
XRD analysis of the mesophase formed by phenyl ether [Ph]c revealed a soft crystalline B phase (SmBcry), which is consistent with a relatively large enthalpy of the isotropic transition (12.7 kJ mol−1) and the optical texture (Fig. 2). The measured layer spacing is 31.2 Å which correlates well with the calculated molecular length of 32.8 Å (including van der Waals radius for H atoms).
Molecular modelling
The energy of coulombic interactions in ion pairs 1c–[Pyr]c was assessed computationally by comparison of the association energy for 1[12]c–[Pyr]c and 2[12]c–[Ph]c for an arbitrary chosen relative orientation of the molecules in the pair. The calculations were conducted using the M06-2x functional, which reasonably well reproduces non-covalent interactions.29Analysis of fully optimized individual structures revealed a nearly linear anion 1[12]c and a bent cation [Pyr]c (Fig. 7). The hexyl chain and the carbonyl group in the former are staggered relative to the {closo-1-CB11} cage. The two carboxyl groups are nearly co-planar with the benzene ring rendering the “organic” half of the molecule planar (Fig. 7a). The angle between the planes of the terminal alkyl chains is 15°, and the molecular length (L, as the H⋯H distance) is 31.6 Å. The carborane analogue 2[12]c has a nearly identical molecular length (L = 31.3 Å). The main difference between 1[12]c and 2[12]c is that the carbonyl group nearly eclipses the B–H bond in the latter, which result in the wider interplanar angle of 27°.
 | ||
| Fig. 7 Equilibrium ground state geometry of ions 1[12]c (a) and [Pyr]c (b) and ion pair 1[12]c–[Pyr]c (c) at the M06-2x/3-21G* level of theory. | ||
The undecyl chain in the pyridnium cation [Pyr]c is orthogonal to the ring plane, which results in a bent molecular structure as shown in Fig. 7b. The interplanar angle is 78° and the H⋯H separation L of 30.3 Å, which is close to the length of anion 1[12]c. Molecular dimensions of the benzene analogue [Ph]c are nearly the same with the interplanar angle of 81° and L = 30.2 Å.
Full geometry optimization of 2[12]c and [Ph]c in vacuum gave a tight molecular pair 2[12]c–[Ph]c, which served as the starting point for geometry optimization of ion pair 1[12]c–[Pyr]c. The resulting close-contact structure is shown in Fig. 7c. A comparison of energies of the individual components and that of the binary system gave the enthalpy of association, ΔHa after BSSE correction. For the non-ionic pair the association is calculated to be mildly exothermic (ΔHa = −4.3 kcal mol−1), while for the ion pair, 1[12]c–[Pyr]c, the association enthalpy is dramatically larger ΔHa = −64.3.0 kcal mol−1 in vacuum. Placing molecules at low strength dielectric medium (ε = 2.5), typical for weakly polar liquid crystals, had little effect on the association enthalpy of 2[12]c–[Ph]c (ΔHa = −3.4 kcal mol−1). In contrast, the dielectric medium significantly reduced the exotherm of formation of ion pair 1[12]c–[Pyr]c (ΔHa = −27.9 kcal mol−1). As a result, the difference in association energy, ΔΔHa, of the non-polar pair and ion pair is 24.5 kcal mol−1 in a weakly dielectric medium. Increasing ε to 10.0, which is reasonable for an ionic liquid,30,31 the ΔΔHa value for the two pairs falls to 7.0 kcal mol−1. Thus, the calculated significant difference in association enthalpy in the two model binary systems is consistent with the observed 181 K higher thermal stability of the mesophase in ILC than in the non-ionic binary mixture. Also, the observed tendency for the formation of high-temperature lamellar phases (SmA and E) by the ion pair instead of a nematic phase is a consequence of the tight molecular arrangement dictated by significant coulombic interactions.
Conclusions
For the first time, we have assessed experimentally the impact of coulombic interactions on mesophase stability by comparison of ionic (1c–[Pyr]c) and isosteric non-ionic (2c–[Ph]c) systems in the absence of other factors. Thus, replacement of appropriate carbon atoms in an equimolar binary mixture with B− and N+ fragments leads to an isosteric and isoelectronic ion pair. Results show that the difference in the clearing temperature between the two binary systems is 181 K, which corresponds to a difference in association enthalpy of 7 kcal in appropriate dielectric media. The boron cluster size in the ionic material appears to have little effect on the magnitude of this phenomenon. It is possible, however, that the size of the ions is the determining factor. A comparison was demonstrated for components of approximately equal length due to their best compatibility of the electrically neutral compounds (i.e. formation of a homogeneous nematic phase).The presented concept of isosteric and isoelectronic replacement of C,C with B−,N+ is applicable to other molecular systems and provides unique experimental models to support theoretical considerations of the liquid crystalline state.
Computational details
Quantum-mechanical calculations were carried out using a Gaussian 09 suite of programs.32 Geometry optimizations for unconstrained conformers of appropriate molecules and ions with the most extended molecular shapes were performed at the M06-2x/3-21G* level of theory29 using default convergence limits. The alkoxy group was set in all-trans conformation maintaining co-planarity with the aromatic ring in the input structure. The aromatic ring and the carboxyl group were staggered with respect to the carborane cage as found experimentally and computationally in related structures. The orientation of the substituents on the cyclohexane ring was set according to results of conformational analysis for model cyclohexane derivatives. Optimized structures of non-polar molecules served as starting points for optimization of the ionic analogues after replacing the carbon atoms with B or N.For calculations of binary system 2[12]c–[Ph]c, molecules of 2[12]c and [Ph]c at equilibrium geometry (M06-2x/3-21G*) were set parallel to each other at a 4 Å distance, and the geometry of the pair was minimized. The resulting energy was corrected for basis set superposition error (BSSE) by running single point calculations (M06-2x/3-21G*//M06-2x/3-21G*) at the equilibrium geometry with the keyword COUNTERPOISE = 2,33 and compared to that of isolated molecular components. The optimized geometry of non-polar pair 2[12]c–[Ph]c served as the starting point for calculations involving ion pair 1[12]c–[Pyr]c, after replacement of the two carbon atoms with B and N. Conformational search was not attempted. For each pair, the association energy was calculated as a difference between the energy of the molecular pair and isolated molecules (ΔHa = Hpair − (Hcat + Han)).
Thermodynamic parameters were requested with the FREQ keyword. The PCM model34 was implemented using the SCRF (Solvent = Generic, Read) keyword and specified “epsinf = 2.25” and “eps = 2.5” or “eps = 10” and the total energies were obtained in single point calculations with the same method.
Experimental part
General
NMR spectra were obtained at 128 MHz (11B) and 400 MHz (1H) in CDCl3 or CD3CN. Chemical shifts were referenced to the solvent (1H) or to an external sample of B(OH)3 in MeOH (11B, δ = 18.1 ppm). Optical microscopy and phase identification were performed using polarized optical microscopy (POM) and a hot stage. Thermal analysis was obtained using a DSC with samples of about 0.5–1.0 mg and a heating rate of 5 K min−1 under a flow of nitrogen gas.General procedure for the preparation of 1–[Pyr]
To a solution of carboxylic acid 4[12]–[NEt4] or 4[10]–[NMe4] (0.1 mmol) in dry CH2Cl2 (2 mL) was added oxalyl chloride (5 eq) and a catalytic amount of DMF. The mixture was stirred at rt for 30 min, and the mixture was evaporated to complete dryness. Dry CH2Cl2 was added (3 mL) followed by phenol 3 (0.1 mmol) and Et3N (0.2 mmol), and the reaction was stirred overnight. Organic products were extracted (CH2Cl2), the organic layer was dried (Na2SO4) and the solvent was evaporated. The residue was passed through a silica gel plug (MeCN/CH2Cl2, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) giving crude product as either the [NMe4] or [NEt4] salt. N-Alkyl-4-alkoxypyridinium bromide ([Pyr]–Br, 1.2 equivalent) was added to a solution of the crude ester 1[10]–[NMe4] or 1[12]–[NEt4] (1 eq) in CH2Cl2 upon which a white precipitate formed. Water was added, and the biphasic system was stirred overnight. The CH2Cl2 layer was separated, and the aqueous layer was extracted with additional CH2Cl2. The CH2Cl2 layers were combined, washed with H2O, dried (Na2SO4), and the solvent was evaporated. The crude product was purified on a silica gel plug (CH2Cl2) and recrystallized from EtOH, EtOH/EtOAc and hexane/EtOAc mixtures providing pure ion pair.
:5) giving crude product as either the [NMe4] or [NEt4] salt. N-Alkyl-4-alkoxypyridinium bromide ([Pyr]–Br, 1.2 equivalent) was added to a solution of the crude ester 1[10]–[NMe4] or 1[12]–[NEt4] (1 eq) in CH2Cl2 upon which a white precipitate formed. Water was added, and the biphasic system was stirred overnight. The CH2Cl2 layer was separated, and the aqueous layer was extracted with additional CH2Cl2. The CH2Cl2 layers were combined, washed with H2O, dried (Na2SO4), and the solvent was evaporated. The crude product was purified on a silica gel plug (CH2Cl2) and recrystallized from EtOH, EtOH/EtOAc and hexane/EtOAc mixtures providing pure ion pair.
General procedure for preparation of esters 2
:1) giving crude ester 2, which was purified further by recrystallization.
:1) giving crude product. The product was further purified by multiple recrystallizations from MeCN/AcOEt and EtOH/AcOEt mixtures giving [Ph]c in 78% yield as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.85 (t, J = 7.0 Hz, 3H), 0.88 (t, J = 6.3 Hz, 3H), 0.90–1.10 (m, 3H), 1.15–1.44 (m, 26H), 1.58–1.66 (m, 2H), 1.68–1.77 (m, 1H), 1.79 (br d, J = 11.8 Hz, 2H), 1.88 (br d, J = 13.3 Hz, 2H), 2.52 (t, J = 7.7 Hz, 2H), 3.72 (d, J = 6.4 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 7.06 (d, J = 8.5 Hz, 2H). Anal. calcd for C29H50O: C, 83.99; H, 12.15. Found: C, 84.19; H, 12.04%.
:1) giving 2.25 g (53% yield) of pyridine 6 as a white solid: mp 72–72 °C; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J = 7.0, 3H), 0.90–1.11 (m, 3H), 1.12–1.36 (m, 10H), 1.70–1.78 (m, 1H), 1.81 (br d, J = 13.8 Hz, 2H), 1.88 (br d, J = 13.8 Hz, 2H), 3.79 (d, J = 6.4 Hz, 2H), 6.78 (d, J = 6.4 Hz, 2H), 8.40 (d, J = 6.4 Hz, 2H). Anal. calcd for C17H27NO: C, 78.11; H, 10.41; N, 5.36. Found: C, 78.15; H, 10.24; N, 5.41%.
Note added after first publication
This article replaces the version published on 15th October 2014, which contained errors in the formulae of the monocarbaborates in the Introduction section.Acknowledgements
This work was supported by the NSF grant DMR-1207585.Notes and references
- T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2008, 130, 1759–1765 CrossRef CAS PubMed.
- T. Ichikawa, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2007, 129, 10662–10663 CrossRef CAS PubMed.
- M. Yoshio, T. Kagata, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2006, 128, 5570–5577 CrossRef CAS PubMed.
- Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco and M. Armand, J. Power Sources, 2006, 154, 255–261 CrossRef CAS PubMed.
- N. Yamanaka, R. Kawano, W. Kubo, T. Kitamura, Y. Wada, M. Watanabe and S. Yanagida, Chem. Commun., 2005, 740–742 RSC.
- R. Kawano, M. K. Nazeeruddin, A. Sato, M. Grätzel and M. Watanabe, Electrochem. Commun., 2007, 9, 1134–1138 CrossRef CAS PubMed.
- Y. Zhao, J. Zhai, J. He, X. Chen, L. Chen, L. Zhang, Y. Tian, L. Jiang and D. Zhu, Chem. Mater., 2008, 20, 6022–6028 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed.
- K. V. Axenov and S. Laschat, Materials, 2011, 4, 206–259 CrossRef CAS PubMed.
- Evidence include induction of liquid crystalline phases by quartenization of the nitrogen center in pyridine and imidazole derivatives. For example: X. Cheng, X. Bai, S. Jing, H. Ebert, M. Prehm and C. Tschierske, Chem.–Eur. J., 2010, 16, 4588–4601 CrossRef CAS PubMed; L. M. Antill, M. M. Neidhardt, J. Kirres, S. Beardsworth, M. Mansueto, A. Baro and S. Laschat, Liq. Cryst., 2014, 41, 976–985 CrossRef.
- D. Demus, in Handbook of Liquid Crystals, ed. D. Demus, J. W. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, New York, 1998, vol. 1, pp. 133–187 Search PubMed.
- S. Kondrat, M. Bier and L. Harnau, J. Chem. Phys., 2010, 132, 184901 CrossRef PubMed.
- G. Saielli, Soft Matter, 2012, 8, 10279–10287 RSC.
- B. Ringstrand, H. Monobe and P. Kaszynski, J. Mater. Chem., 2009, 19, 4805–4812 RSC.
- B. Ringstrand, A. Jankowiak, L. E. Johnson, P. Kaszynski, D. Pociecha and E. Górecka, J. Mater. Chem., 2012, 22, 4874–4880 RSC.
- J. Pecyna, A. Sivaramamoorthy, A. Jankowiak and P. Kaszynski, Liq. Cryst., 2012, 39, 965–971 CrossRef CAS.
- A. Jankowiak, J. Kanazawa, P. Kaszynski, R. Takita and M. Uchiyama, J. Organomet. Chem., 2013, 747, 195–200 CrossRef CAS PubMed.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS.
- Z. Janoušek and P. Kaszynski, Polyhedron, 1999, 18, 3517–3526 CrossRef.
- A. G. Douglass, K. Czuprynski, M. Mierzwa and P. Kaszynski, J. Mater. Chem., 1998, 8, 2391–2398 RSC.
- A. Jankowiak, A. Januszko, B. Ringstrand and P. Kaszynski, Liq. Cryst., 2008, 35, 65–77 CrossRef CAS.
- A. Jankowiak, A. Baliński, J. E. Harvey, K. Mason, A. Januszko, P. Kaszyński, V. G. Young Jr and A. Persoons, J. Mater. Chem. C, 2013, 1, 1144–1159 RSC.
- N. A. Bumagin and E. V. Luzikova, J. Organomet. Chem., 1997, 532, 271–273 CrossRef CAS.
- P. Kaszynski, in Boron Science. New Technologies and Applications, ed. N. S. Hosmane, CRC Press, New York, 2012, pp. 319–354 Search PubMed.
- R. Dabrowski, J. Dziaduszek, J. Szulc, K. Czupryński and B. Sosnowska, Mol. Cryst. Liq. Cryst., 1991, 209, 201–211 CrossRef CAS.
- N. Carr and G. W. Gray, Mol. Cryst. Liq. Cryst., 1985, 124, 27–43 CrossRef CAS.
- For details see the ESI.†.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- T. Singh and A. Kumar, J. Phys. Chem. B, 2008, 112, 12968–12972 CrossRef CAS PubMed.
- M.-M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS PubMed.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS PubMed and references therein.
- G. H. Schmid and A. W. Wolkoff, Can. J. Chem., 1972, 50, 1181–1187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for intermediates 5[12] and 8[11], additional computational data, archive of optimized equilibrium geometries for 1[12]c, 2[12]c, [Pyr]c, [Ph]c, and pairs 1[12]c–[Pyr]c and 2[12]c–[Ph]c. See DOI: 10.1039/c4ra06502f |
| This journal is © The Royal Society of Chemistry 2014 |