
Carbon dioxide as a primary oxidant and a C1 building block†
Abstract
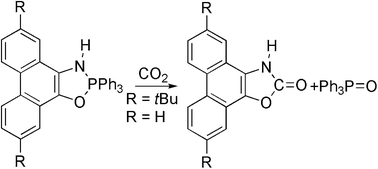
2,3-Dihydro-2,2,2-triphenylphenanthro[9,10-d]-1,3,2-λ5-oxazaphospholes react with carbon dioxide in an overall second order reaction at room temperature to give 3H-phenanthro[9,10-d]oxazol-2-ones and triphenylphosphine oxide in good yields. Nucleophilic attack of the phenolate on CO2 and formation of Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was found.
O was found.
 Please wait while we load your content...
Please wait while we load your content...

