
Polymeric nanocarriers for expected nanomedicine: current challenges and future prospects
B. Daglarab,
E. Ozgurab,
M. E. Cormanc,
L. Uzund and
G. B. Demirel*ae
aUNAM-National Nanotechnology Research Center, Bilkent University, 06800 Ankara, Turkey
bInstitute of Materials Science and Nanotechnology, Bilkent University, 06800 Ankara, Turkey
cDepartment of Biomedical Engineering, Faculty of Engineering, Sinop University, 57000, Sinop, Turkey
dDepartment of Chemistry, Faculty of Science, Hacettepe University, 06800 Beytepe, Ankara, Turkey
eDepartment of Chemistry, Polatli Faculty of Arts and Sciences, Gazi University, 06900 Polatli, Ankara, Turkey. E-mail: gbirlik@gazi.edu.tr
First published on 1st September 2014
Abstract
Polymeric nanocarriers have an increasingly growing potential for clinical applications. The current and future expectation from a polymeric nanocarrier is to exhibit both diagnostic and therapeutic functions. Living organisms are very complex systems and have many challenges for a carrier system such as biocompatibility, biodistribution, side-effects, biological barriers. Therefore, a designed polymeric nanocarrier should possess multifunctional properties to overcome these obstacles towards its target site. However, currently there are few polymeric systems that can be used for both therapy and imaging in clinic studies. In the literature, there are many studies for developing new generation polymeric nanocarriers to obtain future smart and multifunctional nanomedicine. In this review, we discuss the new generation and promising polymeric nanocarriers, which exhibit active targeting, triggered release of contents, and imaging capability for in vivo studies.
1. Introduction
The design of polymeric nanoparticle-based delivery systems has significant impact in biomedical applications combining both therapy and diagnostics. The collaboration of nanotechnology, biotechnology, and polymer science provides a fundamental milestone to construct new diagnostic and therapeutic nanocarriers. Polymeric nanoparticles can provide higher solubility of hydrophobic drugs, increasing drug circulation in blood, and targeted delivery to desired sites compared to conventional therapeutic nanocarriers.1–5 In accordance with these numerous advantages of nanocarriers, medical science today is shifting the focus towards the creation of multifunctional polymeric hybrid systems at the nanoscale. Polymeric nanocarriers (PNCs) are also soft materials and cover micelles, liposomes, dendrimers, and biodegradable and biocompatible polymer-based nanoparticles (Fig. 1).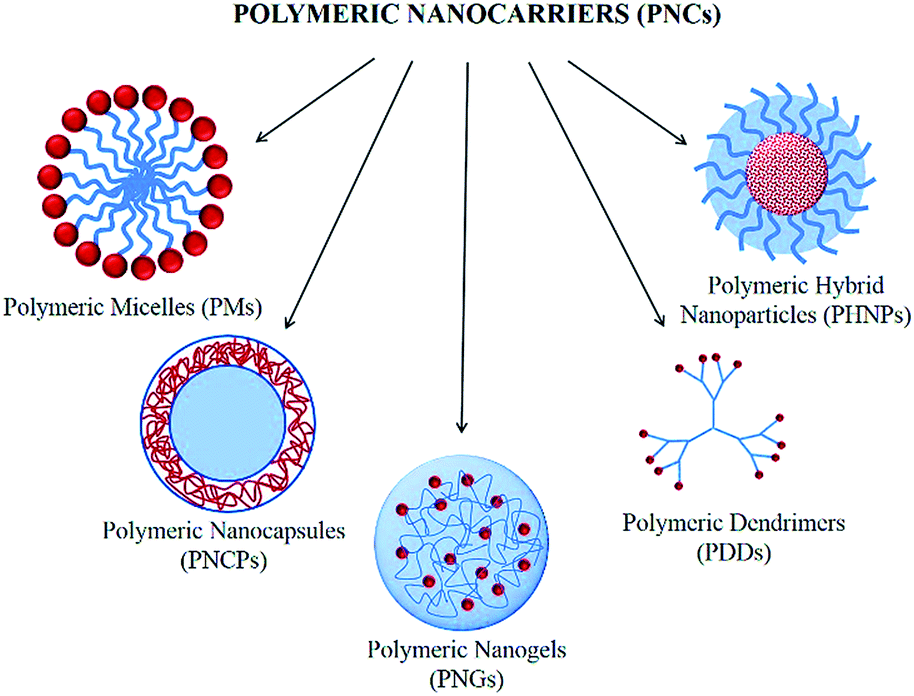 | ||
| Fig. 1 Schematic representation of PNCs. | ||
Polymeric nanocarriers are flexible and biodegradable; therefore, they can be used for controlled release of encapsulated biomolecules such as hydrophilic/hydrophobic drugs, peptides, proteins, DNA, and RNA (Fig. 2).6–8 To fabricate an efficient and multifunctional polymeric nanocarrier system for clinical usage, designed particles should carry certain properties. It is accepted that the size of PNCs plays a major role in determining the in vivo fate of the particles. The size of PNCs is typically between 10 nm and 200 nm to render the biodistribution of particles in the human body.6–8
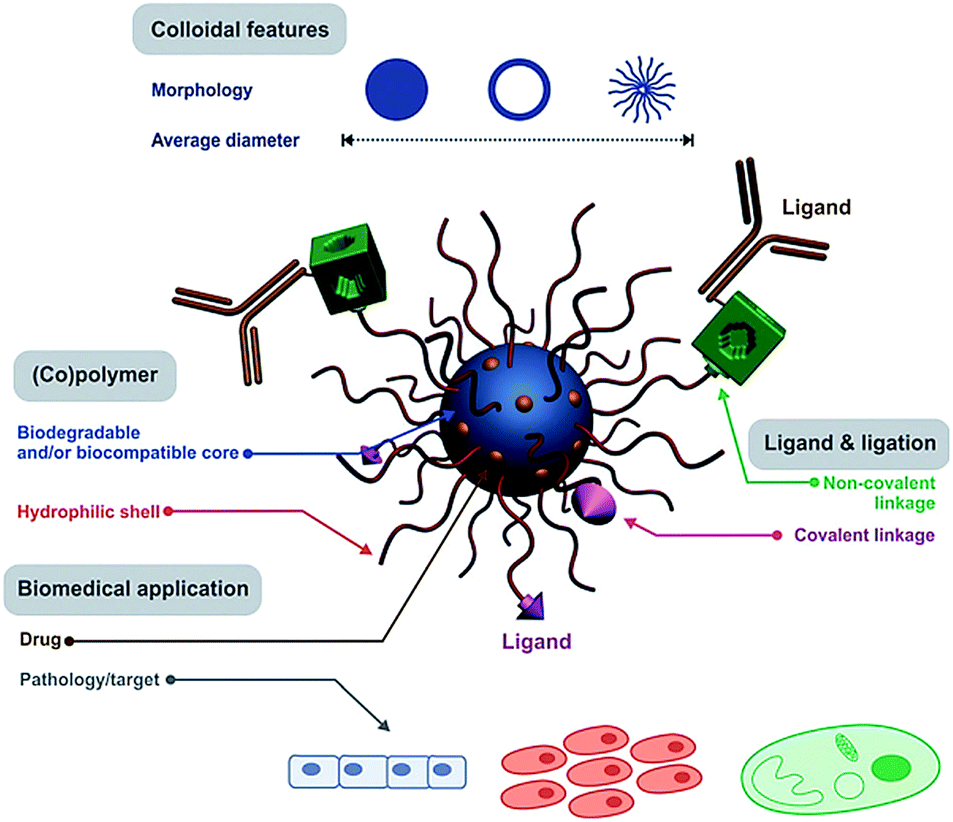 | ||
| Fig. 2 Schematic representation of a polymeric nanocarrier system. Reproduced from ref. 12 with permission from The Royal Society of Chemistry. | ||
The dispersibility and stability of PNCs should not be affected by the changes in pH, ionic strength, polarity or temperature in a physiological or in vivo environment.9–11 In addition to the size of the particles, the surface functionalization is also a considerably significant parameter for clinical applications. Desired nanocarrier systems need to deliver the cargo molecules to the right place, at the right time, and at the right dosage.9–11 In addition to that, PNCs need to have a long circulation time in the bloodstream with a low accumulated toxicity. Consequently, there are many obstacles to overcome in order to achieve a successful drug delivery system. Most of these challenges are related to inappropriate distribution of the cargo molecules, environmental or enzymatic degradation, fast clearance rates, and nonspecific toxicity of PNCs.6–8
Furthermore, PNCs may face numerous barriers on the way to their target, such as mucosal barriers and non-specific uptake. To overcome all the obstacles, the specific properties and the nature of human cell biology should be considered for the design of PNCs.
This review is focused on the scientific progress in polymer-based nanocarriers for theranostic applications. There are still many issues to be considered for PCNs. Therefore, here we will discuss these challenges and also give the recommended solutions with selected state-of-the-art examples.
2. Pharmacokinetic properties of polymeric nanocarriers
2.1. Biodistribution
The distribution of cargo molecules in the human body is a major challenge for delivery systems.10–12 The loaded PNCs must retain the cargo molecules during the transport through the bloodstream. To achieve efficient delivery to the target tissue PNCs must be able to efficiently release the cargo molecules when they reach the targeted tissue, cell or organ.10–12 In this case for successful cargo targeting, the stability and the circulation time of PNCs in the bloodstream are significant parameters. Generally, the circulation process of nanocarriers is expected to be slow. While in slow circulation, the nanocarriers can encounter different obstacles, such as glomerular excretion by the kidney and recognition by the reticuloendothelial system (RES) located in the liver, spleen and lung.9–11For this purpose, the uptake of the cargo loaded PNCs presumably depends on the shape, size, surface charge, and functionalization of the nanocarriers by targeted cells.
2.2. Size and architecture
Size and shape of the nanoparticles have significant influence on the characteristics of PNCs. These parameters can define properties such as drug loading capacity, targeting, accumulation, stability, penetration, and toxicity of nanocarriers. In addition, it is very critical to control nanoparticle uptake by diseased tissue, clearance by the kidney and recognition by the RES.13Cellular uptake and accumulation behaviour of the nanoparticles mostly depends on nanoparticle size, while surface functionalization and surface charge are also important parameters.14 Recently, Kulkarni et al. reported the effect of nanoparticle size (20, 50, 100, 200, and 500 nm) on the gastrointestinal (GI) barrier and the blood–brain barrier (BBB). It is found that the 100 and 200 nm sized polymeric nanoparticles have higher cellular uptake efficiency, and have great potential for drug delivery systems across the GI and the BBB.15
Smaller sized nanoparticles (<200 nm) can escape from the RES without recognition, which results in longer circulation time.14,16 Moreover, the surfaces of the smaller nanoparticles have a small radius of curvature that prevents the binding of opsonins and increases their half-life. Carriers can also be indirectly degraded by the RES. Additionally, bigger nanoparticles (>250 nm) accumulate in different organs, such as liver and spleen, while very small nanoparticles (<5 nm) cannot pass the cut-off limit of the kidney.14,16
In a different approach, polymeric submicellar assemblies are proposed as promising drug carriers compared with the traditional polymeric micelles. These nanostructures are formed below the critical micelle concentration (CMC) of the low molecular weight surfactants. Mendez-Perez et al. reported a PEG-40 stearate (PEG40S) submicellar structure in order to encapsulate lipophilic drugs. Owing to their smaller size than micelles (∼5.8 < ∼14.6), they showed longer circulation time and better permeation.17 This study demonstrates the importance of nanoparticle size even for very slight changes.
There is a huge effort to optimize nanoparticle shape in order to improve controlled drug delivery systems. New carrier architectures are developed to compete with commercial products, which have generally spherical shapes like vesicles. Shape design of the carrier nanoparticles, including branch architecture, significantly affects the penetration, biodistribution, and targeting (Fig. 3). More importantly, nanoparticle shape is another key effect to prevent the clearance of nanocarrier by the RES in addition to the nanoparticle size and surface chemistry. Therefore, new carrier architectures developed during the PNC progress and circulation time of the carriers was increased.18,19 Fox et al. accounted the architectural features of the polymeric drug carriers and their penetration and accumulation abilities to tumour cells.20 It was shown that PEGylated dendrimers and branched polymers have higher lifetime in blood compartment than the globular polymers and well-solvated random coil polymers, while the carriers were rapidly cleared by the liver in the linear polymers case.
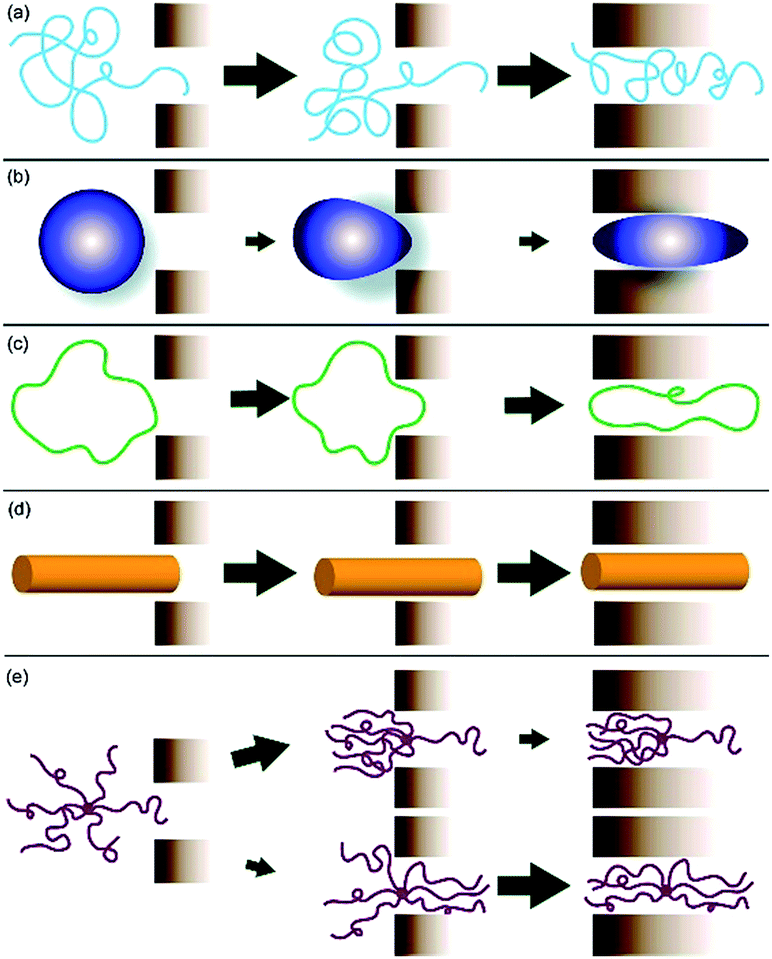 | ||
| Fig. 3 Effect of the polymer shape on cell penetration; (a) linear random coil polymer; (b) polymer with a globular conformation; (c) a cyclic-shaped polymer; (d) tubular-shaped polymer; (e) branched polymers. Reprinted from ref. 20 with permission from ACS Publications. | ||
In addition, De Jesus and Gillies et al. studied the branched structure of PEGylated polyester “bow-tie” dendrimers in order to determine the relationship between branching and blood circulation time.21,22 No significant difference was observed for tissue uptake, while circulation time increased with the degree of branching. Authors attributed this behaviour to the steric hindrance among the polymer and cell pores. In addition to biodistribution and circulation time, carrier architecture also affects the drug loading capacity, release profile of drug, and drug/membrane interactions. Loverde et al. studied the rational coarse grain (rCG) – molecular dynamics (MD) of the interactions between drug and polymeric carrier based on the carrier architecture in detail.23 It is known that worm-like carriers have higher loading and delivery capacity than the spherical carriers and are more effective in tumour shrinkage. Taxol was selected as the drug, which is a common anticancer drug, and carrier materials was selected as poly(ethyleneglycol)-poly(caprolactone) (PEG-PCL). It was determined that free energy of the Taxol is different for worm-like and spherical micelles, and more negative for the worm-likes. Moreover, minimum energy of the Taxol differs for these two architectures and it is close to the interface for worm-like carriers, while it is at the center for spheres. This unexpected distinction can be explained such that it pulls the drug micelle's interface and higher packing constraints for worm-like carriers. In addition, they calculated the change in free energy in PCL core and PCL interface, which is found higher for interface. It was shown that drug loading changes from sphere to worm by two times increment. These findings may explain why worm-like structures function better than the sphericals. It is accepted by the community that increasing the aspect-ratio of polymeric drug carriers increases the circulation time and cellular interactions such as filomicelles, which are nature inspired designs from filoviruses. In addition to these properties, Shuvaev et al. clarified that filomicelles conserve their structural stability after modifying with targeting molecules and informed that antibody-modified filomicelles adhere to endothelial surfaces with a high specificity.24 To reveal the structural stability of the polymeric carriers, the streptavidin–biotin approach was used and carriers were coated with IgG/SA or Ab/SA molecules. I-IgG/SA tracers were mixed to track the carriers and the modified filomicelles bound to endothelial cells, while conserving their shape and size, also anti-PECAM/filomicelles bind specifically compared to control IgG/filomicelles was shown.24 Authors claim that filomicelles are promising for clinical uses owing to their longer retention time in the bloodstream and lower pulmonary uptake levels with minimal non-specific adsorption compared to their counterparts.24
2.3. Surface properties
The stealth properties of nanocarriers can be controlled by their surface properties such as surface chemistry and surface charge. Hydrophilic ligands can prevent the opsonisation, recognition by RES and increase circulation time of the carrier. In addition, proper surface modifications can improve the targeting and penetration abilities of the nanoparticles.12Ornelas-Megiatto et al. examined and compared the effect of phosphonium and ammonium groups on the toxicity and transfection properties of gene delivery systems.25 In addition, the effect of the alkyl substituents was discussed for triethylphosphine, tri-tert-butylphosphine, tris(3-hydroxy-propyl)phosphine, and triphenylphosphine groups. Cytotoxicity experiments of the polymers were studied with cervical cancer cells (HeLa cells) and phosphonium polymer with triethylphosphine terminal group showed 100% cell viability compared to its ammonium analogue. However, while the triethylphosphonium polymer showed high cell viability, it was found out that the alkyl substituents had a significant role in toxicity. Polymer with tri-tert-butylphosphine terminal group was determined to be highly toxic while tris(3-hydroxy-propyl)phosphine polymer did not show a significant toxicity. The binding of the phosphonium and ammine groups to siRNA were also investigated and phosphonium groups presented higher affinity. Authors claim that this difference occurs because of the positive charge position of the terminal groups; the positive charge is centred at the P atom of phosphonium groups, while it is distributed through the adjacent carbons of ammonium groups. Therefore, transfection efficiency for phosphonium groups is 65%, whereas for ammonium groups it is 25%, which shows a significant difference.25
PEGylation is widely used to modify nanocarriers and prevents the removal by RES, hence increases the circulation time.26 However, PEG ligands inhibit the cellular uptake of the nanoparticles. Cleavable PEG ligands are developed to overcome this limitation of the PEGylation by agent L-cysteine. Mei et al. propose a three-tier cascade structure, which consists of RGD, TAT and cleavable PEG.26 Cleavable PEG behaves like a shielding layer and increases the accumulation at tumour tissue; after the cleaving step the nanoparticle can be delivered inside the diseased cells by the synergistic effect of RGD and TAT. TAT allows the delivery of nanocarrier inside the cells with its cationic nature, while RGD supports the specific recognition of diseased tissues. Cleavable PEG, RGD, and TAT have different length of PEG ligands, which is shortened by TAT considering their functions. Different combinations of the PEG, RGD and TAT ligands were prepared to examine theoretical liposome design. Stability, in vitro and in vivo cellular uptake of the nanocarriers have been studied. Findings show that cleavable PEG increases the liposome stability and responds to L-cysteine. Moreover, this three-tier liposome was compared with dual-ligand liposomes and it was shown that internalization of the liposome effectively increased with the synergistic effect of RGD and TAT.26
Layer-by-layer (LbL) deposited poly(lactic-co-glycolic acid) (PLGA) core polymeric carriers were proposed by Morton et al. as an alternative drug carrier system. Polysaccharides were used as antifouling agents to control protein adsorption and to decrease opsonisation effect. Used polysaccharides include different terminated molecules; dextran sulphate (DXS), hyaluronic acid (HA) as a cancer cell receptor and alginate (Alg) as a protective layer to evade immune system. Differently coated PLGA core nanoparticles were compared with each other and with the uncoated nanoparticles. In vivo and in vitro studies showed that Alg and HA including nanocarriers significantly increased the drug half-life and minimized the drug accumulation in the liver. Obtained results indicate that LbL nanocarriers are potential candidates for controlled drug release systems.27
Peng et al. reported polymeric nanoparticles (poly-3-hydroxybutyrate-co-3-hydroxyhexanoate (PHBHHx)) with albumin corona to prevent opsonisation and rapid clearance from the blood. Bovine serum albumin (BSA) coated and uncoated polymeric nanoparticles were compared to understand non-specific interactions with immunoglobulin (IgG), which is an important opsonin, and observed that BSA corona inhibits the IgG adsorption onto the nanoparticles and results in a lower opsonisation. In addition, cytotoxicity experiments showed that albumin coated nanoparticles are less toxic than the uncoated nanoparticles especially at higher concentrations. In addition to these improvements, a significant increase is observed in the size and zeta potential of the BSA coated nanoparticles. Although size increase with the BSA coating does not influence the biodistribution of this system, this increase is discussable for other polymeric nanoparticles and uses.28
To increase the stealth properties of polymeric nanoparticles, polylactic acid (PLA)-based nanocarriers were studied by Sheng et al. Different surface coatings, such as water soluble cationic partially deacetylated chitin (PDC), anionic N-carboxy propionyl chitosan sodium (CPCTS), and their combinations with PEG were investigated. They showed that PEG/PDC combined nanoparticles have significantly higher half-life in blood circulation than their single coatings. In addition, biodistribution of the nanoparticles was determined in vivo and found that small amounts of nanoparticles isolated by the liver. Authors claim that the combination of PEG/PDC coating can be an effective alternative to their single coatings to increase the circulation time of the polymeric drug carriers.29
Another approach not to be recognized by the RES and to increase the circulation time of the nanocarrier is to modify the surface of nanoparticles with negatively charged molecules. However, circulation time and cellular internalization requirements work in reverse. Because nanocarriers should be charged positively for cellular internalization, dual pH-responsive systems are developed. While the pH value of normal tissue is ∼7.4, it is ∼6.8 for tumour extracellular environment. Lv et al. proposed a smart polyionic system that is composed of an anionic methoxy poly(ethylene glycol)-b-poly(L-glutamic acid-co-L-phenylalanine) (mPEG-b-bP(Glu-co-Phe)) copolymer and a cationic methoxy poly(ethylene glycol)-b-poly(L-lysine-co-L-phenylalanine) (mPEG-b-P(Lys-co-Phe)) copolymer in order to deliver an anti-tumour drug doxorubicin (DOX).30 Polymeric complex is negatively charged above pH 7.0 and positively charged at pH 6.8, which is convenient for charge conversion expectations. DOX loaded nanoparticles showed different release behaviour at different pH values within the same period, 30% of DOX was released at pH 6.8 and 65% of DOX was released at pH 5.0, while only 17% was released at pH 7.4, indicating these nanocarriers are also endo/lysosomal pH-responsive. In vivo maximum tolerated dose (MTD) and anti-tumour studies showed that DOX loaded nanoparticles (DOX-NPs) were safer and represented higher anti-tumour efficacy than DOX free nanoparticles. This behaviour was attributed to the slow release kinetics, long circulation time, and enhanced cellular uptake of DOX-NPs.
There are various drug carrier systems with different terminated groups such as single coating, LbL or three-tier liposomes and ammonium, phosphonium, PEG, albumin, or polysaccharides. It is very difficult to compare these systems with each other, but their comparison in their self-studies is more reliable. PEGylation increases the blood circulation time of the nanocarriers, however it inhibits the cellular uptake of the drug carriers. To overcome this disadvantage of the PEGylated particles, cleavable PEG systems were developed. It is obvious that phosphonium groups increase the transfection efficiency more than the ammonium groups. In addition, multi-layer nanoparticle systems were proposed to increase the half-life of nanoparticles and minimize the opsonisation. Developed systems are promising for therapeutics and still open for further innovative designs.
2.4. Biological barriers
A wide range of biological barriers exist to protect the human body from invasion by foreign materials.31,32 Skin, muscle, cellular and mucosal barriers can be considered among others. The nanocarriers need to pass these barriers and reach the blood circulation in order to access the target site for an efficient therapeutic efficacy. Among different types of nanoparticles systems, such as inorganic, liposomes, and micelles, polymeric nanoparticles have a great promising potential in using nanocarriers for therapeutics because of their soft and easily functionalizable nature. Moreover, synthetic biodegradable or biocompatible polymers are preferred because of their predictable chemical and physical properties, such as solubility and permeability. The type of barrier may change with the route of administration. Thus, the distribution of PNCs may be different through different routes, and the physical and chemical properties of PNCs might have a strong influence on the therapeutic outcome.The first barrier; i.e., skin, is mostly explored for the local delivery of nanoparticles.33 Nanocarriers' delivery to epidermis, which is the deeper layer of the skin, without barrier modifications was achieved with a little success because of the multilayer nature of the skin.33 Michinaka and Mitragotri demonstrated the feasibility of the injecting polymeric particles into skin using needle-free liquid jet injectors for skin delivery of the loaded therapeutic agent.33
In a recent study, Wang et al. prepared new cationic TAT-conjugated polymeric lipid vesicles (TPLVs) formed from amphiphilic lysine–linoleic acid modified dextran (LLD) and cholesterol (Chol) to be used as transdermal drug delivery carriers.34 To visualize the skin penetration of TPLVs in vivo, they used different particles (Fig. 4). To compare the skin penetration of TPLVs with other particles, they also used conventional liposomes (CLs) and lipid vesicles (PLVs). They modified the entire particle with a common hydrophilic fluorescence probe, calcein35 to visualize the situations into skin layer. They observed that the encapsulation into liposomes clearly enhances the penetration of calcein. For intravenous processes, PNCs reach the targeted site through blood circulation via other routes. This suggests different barriers and circumstances need to be considered for PNCs. First, the particle size of PNCs needs to be around 200 nm to escape from the complex mechanisms of the spleen.36 On the other hand, if the particles are larger than 200 nm, they need to be modified with hydrophilic and biocompatible agents in order to prevent their accumulation and ensure they would remain within the blood circulation for prolonged periods of time. As a general principle, biocompatible agent modified nanocarriers demonstrate unique medical effects depending on their structure. They can cross biological barriers and cellular membranes and interact with cellular receptors. Cell penetrating peptide modified multifunctional NCs can cross cell barriers and become preferably retained within target cells via the endowed permeability and retention (EPR) effect.37,38 Jang et al. prepared a conjugate (DA3) of deoxycholic acid and low molecular weight polyethylenimine (PEI), which has a property that mimics the properties of the cell penetrating peptides (CPPs), micelle-like core–shell PNCs for simultaneous delivery of an anticancer drug and siRNA. They demonstrated that the drug-loaded cationic micelles can then interact with siRNA to form stable complexes. This stable micelle-like complex showed significantly enhanced inhibition of the cancer cell growth into tumour-bearing animals.39 Kong et al. developed FITC labelled-polyamidoamine (PAMAM)-based dendrimers for targeting non-small cell lung cancer. They used polymeric dendrimers because dendrimers are a type of branched polymers. These polymers have a large number of functional groups and allow binding of multiple biological molecules. These polymeric dendrimers were efficiently taken up by the non-small lung cancer cells and tumours.40
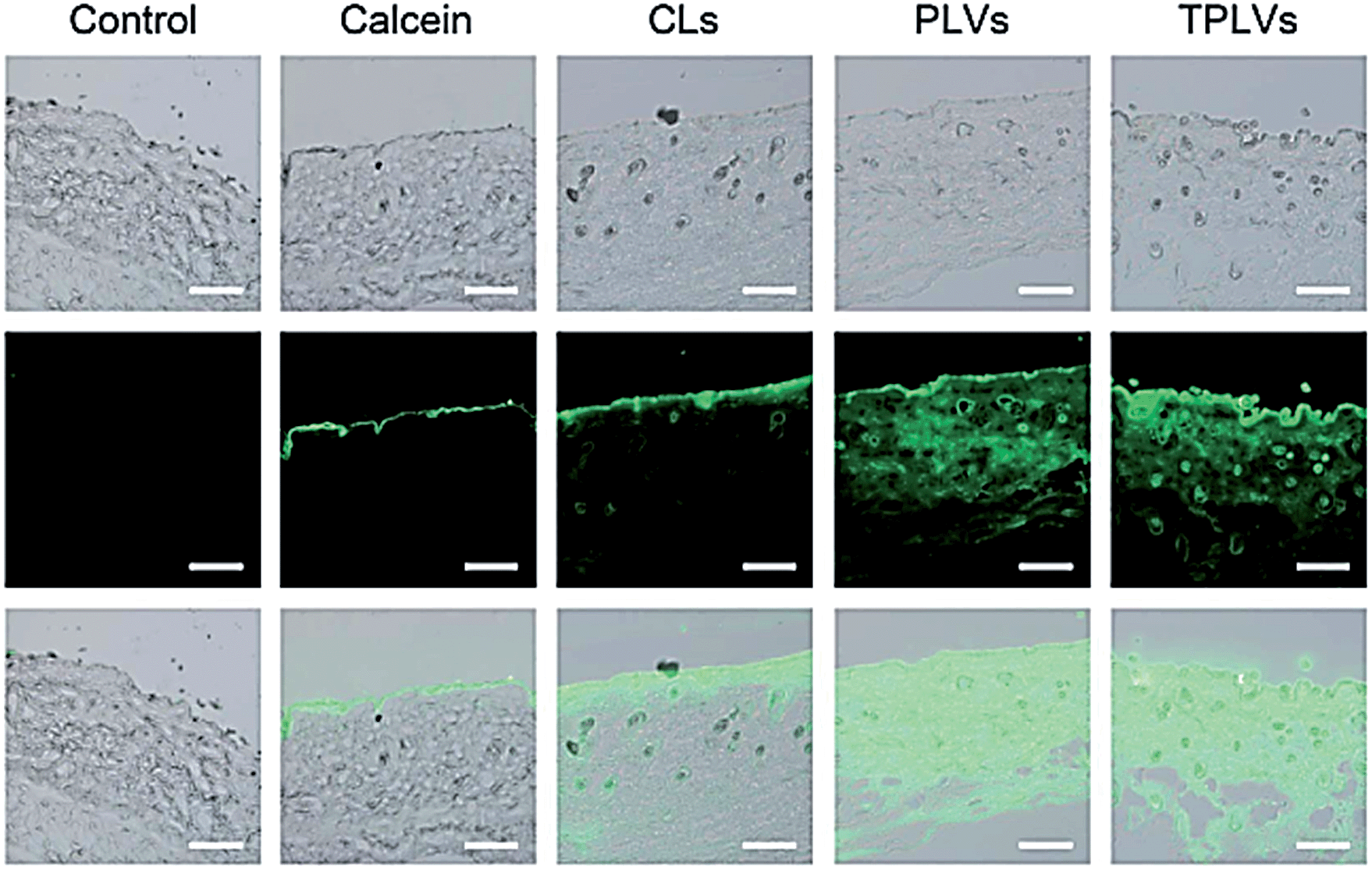 | ||
| Fig. 4 In vivo mouse skin permeation of the samples of TPLVs. Reproduced from ref. 34 with permission from The Royal Society of Chemistry. | ||
Biodegradable and biocompatible polymer nanoparticles are enormously used for nano-therapeutics because of their multifunctional properties. Poly(D,L-lactic-co-glycolic acid) (PLGA) and polylactic acid (PLA) have been widely used for a variety of biological applications because of their biocompatibility. Saltzman et al. investigated the entrapment efficiency of rhodamine-loaded PLGA NPs in three different epithelial cell lines modelling the respiratory airway (HBE), gut (Caco-2), and renal proximal tubule (OK). They observed that the rates of uptake of both the Caco-2 and the HBE cells were considerably slower than in the OK cells. The authors showed that PNCs trafficking can be used for further efforts in targeted PNCs applications, in which cell recognition and association with cell membranes can be enhanced by the use of surface ligands to bind specific receptors on cell membranes.41 Another most challenging barrier can be defined as blood–brain barrier (BBB). The responsible barrier is the capillaries of the brain, and these capillary endothelial cells are characterized by having tight continuous circumferential junctions that control and limit the access of molecules. Many therapeutic drugs cannot access the brain because of the BBB. In this case nanocarriers have promising potential to overcome this problem, while improving drug targeting, reducing drug toxicity, and improving therapeutic efficacy.42,43 In a recent study, Sabel et al. investigated the effect of the physical properties of PNCs such as size, charge and the presence of surfactant, for allowing BBB passage.44 They synthesized various fluorescent labelled polybutylcyanoacrylate (PBCA) nanocarriers by mini-emulsion polymerization by changing charge, size, and surfactant compositions. They imaged the PNCs passing over the blood–retina barrier (a model of the BBB in live animals) as seen in Fig. 5. According to their experiments, they reported that the size and charge of PNCs had no influence on BBB passage and cell labelling. Thus, neither NP's size nor chemo-electric charge, but particle surface is the key factor determining BBB passage. It was observed that PNCs can serve one of two opposite functions: while non-ionic surfactant enhances brain up-take, addition of anionic surfactant prevents it.
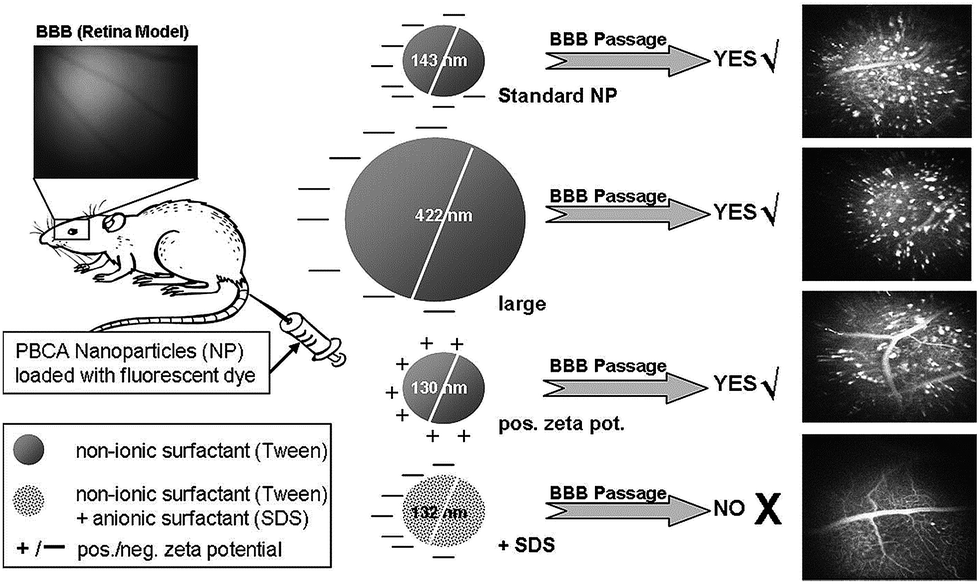 | ||
| Fig. 5 Surfactants, neither size nor zeta-potential influence blood–brain barrier passage of polymeric nanoparticles. Reprinted from ref. 44 with permission from Elsevier. | ||
They proposed that PNCs engineering for therapeutic agents for BBB passers depends on the surfactant composition and PNCs can be designed to specifically enhance drug delivery to the brain; alternatively, to prevent brain penetration such that to reduce unwanted psychoactive effects of drugs or prevent environmental nanoparticles from entering tissue of the central nervous system.44
2.5. Cytotoxicity
Cytotoxicity is defined as the toxic effect of materials on viable cells. It is among the main concerns regarding the production of nanoparticles. Although cytotoxicity is desired in many cases, sometimes biocompatibility is more important. In order to reveal these properties of nanoparticles, several commercial cell lines could be used to draw a projection for potential applications of nanoparticles. Generally, nanoparticles are added into the cell-culture and proliferation and differentiation of the cells are monitored in addition to control cell-culture, used for comparison. To monitor the cells cultured in presence/absence of the nanoparticles, flow cytometry, microscopies including confocal, optical, fluorescent, scanning electron and transmission electron, cell viability and cell cycle analyses, and microplate/ELISA readers, could be used. Data obtained are generally assessed through methylthiazoltetrazolium (MTT) assay to discuss the efficiency of nanoparticles developed. The incubation time is also varied from a few hours up to 72 h to evaluate efficient culture period along with some external effects such as magnetic field and light emission on cell-culture. Cell-lines are selected with respect to targeted cancer type, i.e. breast, kidney, lung, murine, and epithelial. Rosenholm et al. used HeLa cell line to evaluate the efficiency of porous silica hybrid nanoparticles.45 They used flow cytometry and confocal microscopy for monitoring cells while varying the concentration of nanoparticles and incubation time (up to 72 h). They reported that the cell uptake was observed within 2–3 h, but efficient cytotoxicity was observed in 24 h. Cells undergo apoptosis pathway depending on the evidences on gross changes in their nuclear structure. Kocbek et al. used T47-D breast cancer cells by using scanning and transmission electron microscopies, fluorescent microscopy instruments and cell viability and cell-cycle analyses.46 They performed cytotoxicity analysis whether in presence or absence of external magnetic field. They reported that external magnetic field resulted in drastic changes in cellular metabolic activity at a concentration of 100 μM mL−1 within 24 h. Zhang et al. used HepG2 cells for cytotoxicity test, while using microplate readers to collect data and applied MTT assay for cell-proliferation monitoring.47 They reported that cytotoxicity of nanoparticles mainly depends on their formulations. At the highest nanoparticle concentration (400 mg L−1) used, the percentage of viable cells was higher than 95.0%. The IC50 value for nanoparticles changed with respect to incubation time, varying from 24 h to 48 h. They reported cytotoxicity of nanoparticles for HepG2 cells was lower and increased slightly in accordance to free active agent (doxorubicin). Gu et al. used MCF-7 and HUVECs as targeting cell lines via applying MTT assay on ELISA reader data.48 They reported that the cell viability was in the range of 99.3–102.5% and 99.0–103.7% for MCF-7 and HUVECs, respectively. The results showed that the nanoparticles developed had almost no cytotoxicity on those cell-lines up to the nanoparticle concentration of 10 μg mL−1. However, cell viability decreased in a dose-dependent manner. They also reported that a gradual decrease in the cell viability occurred with increase in incubation time. Bailly et al. used MCF12A and MDR-MDA-MB-231 breast cancer epithelial cells for evaluating efficiency of the block polymer cytotoxicity.49 They reported that the nanoparticles had no cytotoxicity in the concentration range of 10–1000 μg mL−1; however, incorporation of hydrophobic drug (clofazimine) decreased the viabilities of the cells. Liu et al. used Lewis lung carcinoma (LLC) and glioma 9L cells for evaluating cytotoxicity of the camptothecin carrying nanoparticles.50 They reported that unloaded nanoparticles had no toxic effect even at high concentration of drug (up to 3 mg mL−1). On the other hand, camptothecin loaded nanoparticles had a significant cytotoxicity even at low drug concentration of <1 μM. Chan et al. used both HeLa and HepG2 cell lines for assessing cytotoxicity of core–shell nanoparticles.51 They also reported that plain nanoparticles did not cause a significant cytotoxicity against cell lines, suggesting low in vitro cytotoxicity up to 300 mg kg−1 body weight doses. Tan et al. used MCF-7 breast cancer cells to show the cytotoxicity of quantum dots/iron oxides incorporated into nanoparticles.52 They reported that the cell viability for quantum dots/iron oxides incorporated nanoparticles (95.4%) was better than that for free quantum dots (81.3%) and that for free iron oxides (80.5%). They emphasized that incubation time adjusted as 24 h and 48 h also affected cell viability, in which increase in incubation time caused a decrease in cell mortality. Ma et al. used A549, HepG2, MCF-7 and C26 cell lines for evaluating cytotoxicity of hydrophobic drug (paclitaxel) carrier nanoparticles.53 They reported that no significant cytotoxicity was observed for all cell species although cytotoxic effect had been reported with high concentration up to 7821.5 ng mL−1. Tian et al. used HeLa cell lines while evaluating near-infrared photodynamic therapy application of cell specific rubyrin-based component incorporated nanoparticles.54 Cytotoxicity was monitored in presence or absence of light irradiation at 635 nm. They reported that nanoparticles showed high phototoxicity under irradiation of 30 J cm−2, although no cytotoxicity was observed in absence of light emission. Moreover, the cytotoxicity increased with increasing nanoparticle amount, having half lethal dose (IC50) of 35 μg mL−1 over 4 h of incubation period. Gong et al. used the HEK293 cell line for evaluating the cytotoxicity of the hydrophobic drug (honokiol) carrier biodegradable micelle nanoparticles.55 They performed the evaluation at 24 h and 48 h incubation time, while varying nanoparticle concentration in the range of 0–2000 μg mL−1. At the highest nanoparticle concentration (2000 μg mL−1), the cell viability decreased from 96.2% to 80.5% by increasing the incubation time from 24 h to 48 h. They mentioned that the nanoparticles developed were biocompatible with very low cytotoxicity and could be classified as safe drug carriers.3. Loading in polymeric nanocarriers
The main challenges in cancer therapy are the delivery of the active therapeutic agents directly to cancer cells and adjusting the release kinetics of these agents from their carrier system, because most of the commonly used anticancer drugs have serious side-effects on not only cancer cells, but also healthy cells and tissues, which interact with these agents during transportation to the target. In addition, because of short half-life and low solubility, especially for hydrophobic drugs, the agents are injected in doses higher than required.1,12 The release of these chemotherapeutic agents is being controlled because of burst effect on the releasing sites. Therefore, different approaches have been developed to improve the releasing dynamics at only targeted sites.1,12 The simplest method to load cargo molecules onto PNCs is physical adsorption. Reddy et al. prepared magnetic polyacrylamide/gelatine nanocomposites to carry doxorubicin that is commonly used in the treatment of a wide range of cancers, such as haematological malignancies and soft tissue sarcomas.56 They performed physical adsorption to load the drug by simply immersing the nanocomposites in the drug solution. They reported the loading efficiency values in the range of 48–64% in respect to nanocomposite formulation. They also reported sustained and improved release dynamics because of the alignment of magnetic nanocomposites by the external magnetic field applied.Dialysis is another simple loading approach to form drug-loaded micelles. Gu et al. reported amphiphilic polymers to load docetaxel by dialysis in the presence of dimethylformamide as a solvent and phosphate buffered saline (PBS, 10 mM, pH 5.5) as a dispersion solution.48 After micelle formation, dialysis membrane having cut-off as 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was used to separate the drug-loaded micelles. They determined the drug loading efficiency by using high performance liquid chromatography via distributing them in tetrahydrofuran and reported drug loading content in the range of 0.32–11.2% and drug loading efficiency in the range of 1.3–67.2% in regards to the formulations applied. Bailly et al. also used a dialysis method to load clofazimine onto polyvinylpyrrolidone-based polymer aggregates.49 Drug and polymers were dissolved in water miscible organic phase, dimethylsulfoxide, and dialyzed against distilled water. They reported that the increase in the hydrophobic blocks in polymeric aggregates caused the increase in drug loading and encapsulation efficiencies. They reported that the loading capacity values were in the range of 1.4–20.0% by weight, whereas the encapsulation efficiencies were in the range of 13–60% by weight in respect to drug in-feeding amount. Chan et al. also used dialysis to prepare docetaxel loaded nanoprecipitate based on poly(lactide-glycolide)/poly(ethylene glycol) nanoparticles.51 They reported that drug loading and encapsulation efficiencies were about 31–32 μg mL−1 and 62%, respectively, with initial polymer concentration of 0.5 mg mL−1 and docetaxel concentration of 50 μg mL−1.
000 was used to separate the drug-loaded micelles. They determined the drug loading efficiency by using high performance liquid chromatography via distributing them in tetrahydrofuran and reported drug loading content in the range of 0.32–11.2% and drug loading efficiency in the range of 1.3–67.2% in regards to the formulations applied. Bailly et al. also used a dialysis method to load clofazimine onto polyvinylpyrrolidone-based polymer aggregates.49 Drug and polymers were dissolved in water miscible organic phase, dimethylsulfoxide, and dialyzed against distilled water. They reported that the increase in the hydrophobic blocks in polymeric aggregates caused the increase in drug loading and encapsulation efficiencies. They reported that the loading capacity values were in the range of 1.4–20.0% by weight, whereas the encapsulation efficiencies were in the range of 13–60% by weight in respect to drug in-feeding amount. Chan et al. also used dialysis to prepare docetaxel loaded nanoprecipitate based on poly(lactide-glycolide)/poly(ethylene glycol) nanoparticles.51 They reported that drug loading and encapsulation efficiencies were about 31–32 μg mL−1 and 62%, respectively, with initial polymer concentration of 0.5 mg mL−1 and docetaxel concentration of 50 μg mL−1.
Liu et al. used a modified oil-in-water single emulsion technique to produce (S)-camptothecin (CPT)-loaded nanoparticles based on the carriers including lactone/succinate functionalities.50 Polyvinyl alcohol was used as a stabilizer of emulsion, while methylene chloride and TMX400 sonic disruptor were used as a solvent and emulsion former. They reported that drug loading ranged from 12% to 22%, while encapsulation efficiency was close to 95%. Ma et al. developed a biocompatible block copolymer for carrying a hydrophobic drug, paclitaxel, through single emulsion solvent evaporation. They reported optimal drug entrapment efficiency and drug loading as 38.02% ± 4.51% and 93.90% ± 4.56%, respectively.53
Tian et al. reported cell-specific and pH-activatable nanoparticles for rubyrin release through single step sonication method. Subsequent to the synthesis of nanoparticles, they determined that drug encapsulation efficiency measured by UV-vis spectroscopy was 48.8% in which nanoparticles have a half lethal dose (IC50) of 35 μg mL−1 at incubation of 4 h under photo-initiated therapy.54 Gong et al. developed biodegradable self-assembled poly(ethylene glycol)/poly(caprolactone)-based micelles for hydrophobic honokiol delivery through ultrasound dissolution method.55 They measured honokiol concentration and loading efficiency using high performance liquid chromatography equipped with reversed phase C18 column. They reported that drug loading was 6.7% when applying in-feed mass ratio of honokiol/copolymer as 1/5. The particles had average particle size, polydispersity index, and zeta potential around 58 nm, 0.266, and −0.4 mV, respectively.
Another approach for drug loading to control drug delivery efficiency is the core–shell entrapment method. Zhang et al. developed self-assembled pH-responsive block copolymer micelles for the delivery of anticancer drug, doxorubicin, through the core–shell entrapment method.47 These copolymers having poly(ethylene glycol) methyl ether, poly(lactic acid), and poly(amino acid ester) segments directly self-assemble into core–shell micelles in aqueous solution at low concentration. They reported that actual doxorubicin loading efficiency was in the range of 18.28–22.00% in regards to sub-segment composition. They also reported that drug release/loading efficiency directly depended on the pH-change in the media. Cho et al. reported a core–shell entrapment method for developing a multifunctional nanocarrier system for cancer diagnosis and treatment.57 They used quantum dots with emissions in the near-infrared range (∼800 nm) as core material and decorated them with poly(styrene) matrix (∼150 nm) consisting superparamagnetic iron oxide (Fe3O4) nanoparticles (∼10 nm). They demonstrated the drug loading efficiency using paclitaxel as a model drug. They reported that drug concentration associated with 50% inhibition of the mitochondrial enzyme activity (IC50) was 125 ng mL−1.
Chemical immobilization of drug molecules onto a carrier system is one of the safety approaches to control harmful side-effect of the drugs through delivery path and onto healthy cells. Messerschmidt et al. developed targeted lipid-coated polymeric nanoparticles displaying tumour necrosis factor on their surface.58 In this manner, they aimed to activate tumour necrosis factor receptor 1 and 2, which led to enhance cellular apoptosis. In this strategy, they performed multi-step modification in which they started with amino-functionalized latex. The amino groups of the nanoparticles were activated with 3 mM sulfosuccinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMMC). Then, they immobilized a cysteine functionalized derivative of single chain tumour necrosis factor (cys-scTNF) onto activated nanoparticles. Using this multi-step modification procedure, they developed nanocarrier systems diminishing non-specific adsorption onto healthy mammalian cells, controlling selective delivery to antigen-positive cells, reducing off-target cytotoxicity, and verifying the effective shielding of tumour necrosis factor activity. Ponta and Bae also performed the chemical immobilization method to develop a tunable drug carrier system.59 For this purpose, they synthesized poly(ethylene glycol)-β-benzyl-L-aspartate block copolymers through ring opening polymerization. Then, they introduced carbazate drug-binding linkers via modification of the side chains of aspartate functionalities. Then, the ester groups were converted into hydrazine through aminolysis reaction with hydrazide molecules. Finally, a chemotherapeutic agent, doxorubicin, was immobilized onto the amino groups of hydrazone functionalities incorporated into aspartate side-chains. In this step, the reaction conditions for doxorubicin conjugation were optimized through the extensive variation of effective factors such as solvent temperature and concentration. They measured drug loading of the final product by using UV-vis spectroscopy at 480 nm, which was in the range of 2.8–32% in respect to weight percent.
In conclusion, several drug loading approaches were developed for enhancing drug loading capacity, while controlling/diminishing harmful side-effects of anticancer drugs against not only targeted tumour cells, but also tissues in the delivery pathway and healthy mammalian cells.
4. Targeted cargo release
It is possible to achieve functionalization of nanoparticles for cancer treatment through two different ways: starting with functional (bio) molecules to synthesize the nanoparticles, or activation and ligand immobilization after nanoparticles synthesis. In the literature, several nanoparticle platforms were reported by these approaches. Although it is hard to classify these materials into a single simple chart, the main structure, functionalization, targeting bio/ligand, and modifier could be used for classification (Table 1). Rosenholm et al. prepared hybrid silica nanoparticles via hyperbranching polymerization on porous silica nanoparticles.45 Hyperbranching was achieved by polyethyleneimine, which was used as a modifier and for increasing surface functionality to immobilize the bioligand at the appropriate amount. In that study, they used folic acid as targeting bioligand for cancer cells. Kocbek et al. proposed superparamagnetic poly(lactide-co-glycolic acid) nanoparticles for targeting intracellular compartments.46 They used ricinoleic acid to hydrophobize superparamagnetic iron oxides. Messerschmidt et al. reported combinations of tailor-made complex polymeric nanoparticles with liposomes (immunoliposomes) to generate multifunctional lipid–polystyrene nanocomposite systems.58 Cancer cell targeting ability was gained by both single chain TNF functionalization on the polymer surface and single-chain Fv–polyethylene glycol–lipid insertion into lipid shell. Zhang et al. reported block copolymer micelles having pH-responding ability.47 In that study, researchers combined hydrophilic (polyethylene glycol methyl ether), hydrophobic (polylactic acid), and pH responsive (poly-β-amino esters) into block copolymer forms including doxorubicin as model drug. Fang et al. also reported lipid–polymer nanocomposite systems including lactide and glycolic acid as end functional groups and polyethylene glycol and lecithin as modifiers through easy single step sonication of component cocktails in different compositions.60 Gu et al. reported di-block copolymers as a main structure, while using racemic amino acids (D,L-leucine) as targeting ligand. They applied ring opening polymerization of racemic-N-carboxy-leucine anhydrides.48 Bailly et al. also reported block copolymers for hydrophobic drug delivery. They used polyvinyl acetate and poly(N-vinylpyrrolidone) as starting polymers for blocking them into a single polymeric nanoparticle chain.49 For this aim, they previously synthesized macro RAFT agent based on polyvinylpyrrolidone by using S-(2-cyano-2-propyl)-ortho-ethyl xanthate as RAFT initiator. Tong et al. reported photoswitchable lipid–polymer nanoparticles including photo-conversion of spiropyran to merocyanine.61 They modified spiropyran groups with hydrophobic alkyl chain and combined them with different polyethylene glycol-based lipids and lecithin to obtain nanoparticles with different chemical and physical properties. Liu et al. reported polyester nanoparticles including 1,4-butanediol as substrate for enzymatic hydrolysis by lipases of terpolymers obtained from ω-pentadecalactone, butylene, and succinate sub-chains.50 Chan et al. reported core–shell nanoparticles produced through modified nanoprecipitation.51 They used soybean lecithin as modifier and poly(lactide-co-glycolic acid) as core material with polyethylene glycol as end functional group (shell) for improving cell-penetration and biocompatibility. Tan et al. also reported copolymer nanoparticles containing iron oxides and quantum dots as agents for multimodal tumour imaging studies.52 They also applied a modified nanoprecipitation method to obtain the desired nanoparticle system in which lactic acid and polyethylene glycol 1000 succinate groups formed surface functionalities. Cho et al. also reported multifunctional nanoparticle systems including fluorescent and superparamagnetic nanospheres for not only drug storage, but also for targeting and imaging of cancer cells.57 For these aims, they immobilized quantum dots onto polystyrene nanoparticles embedded with iron oxide nanoparticles. Cell targeting was achieved by immobilization of anti-prostate specific membrane antigen as bioligand, whereas paclitaxel was used as the model drug. Ma et al. reported hydrophobic nanoparticles including cholesterol as targeting agent and L-glutamate groups as modifier.53 The main skeletons of the nanoparticles were formed by biocompatible poly-ethylene glycol chains. Tian et al. reported nanoparticles with both pH- and photo-responsive abilities, which were prepared through a single-step sonication method.54 Introduction of selenium into rubyrin core and dimethylaminophenyl moiety at the meso-position of rubyrin gained oxygen generation ability and pH-controllable activity, respectively. They also used folic acid as bioligand for cancer cell targeting via folic acid–folic acid receptor interactions. As seen in this short literature survey, the researchers reported different modification strategies to achieve specific cancer cell targeting, while limiting the adverse effects of high drug dose and improving biocompatibility and cellular uptake of proposed particulate systems.| Main structure | Functionalization | Modifier | Targeting (bio)ligand | Ref. |
|---|---|---|---|---|
| Hybrid silica | Hyperbranching polymerization | Polyethyleneimine | Folic acid | 45 |
| Poly(lactide-co-glycolic acid) | Superparamagnetic iron oxides | Ricinoleic acid | Lactide and glycolic acid | 46 |
| Lipid-PEG nanocomposites | Lipid layers | Single chain TNF functionalized surface | Antibody against fibroblast activation protein | 58 |
| Block polymers | Methyl ether poly(ethylene glycol) | Acryloyl chloride | Lactic acid and β-amino acids | 47 |
| Lipid-PEG nanocomposites | Sonication of cocktail including predetermined component concentration | PEG/lecithin | Lactide and glycolic acid | 60 |
| Di-block copolymers | Ring opening polymerization | Racemic-N-carboxy-leucine anhydride | Leucine and ethylene glycol | 48 |
| Block copolymers | RAFT | PVP macro-RAFT agent | Acetate/pyrrolidone | 49 |
| Photoswitchable nanoparticles | Spiropyran with hydrophobic alkyl chain | Lecithin/polyethylene glycol | Cell penetration peptide (CPP) (Cys-Tzt) | 61 |
| Polyester nanoparticles | Ter-polymerization | Novozyme 435 | 1,4-Butanediol as substrate for lipases | 50 |
| Core–shell nanoparticles | Modified nanoprecipitation combined with self-assembly | Soybean lecithin | Polyethylene glycol | 51 |
| Copolymer nanoparticles | Modified nanoprecipitation combined with quantum dots and iron oxides | Polylactic acid | Tocopheryl polyethylene glycol succinate | 52 |
| Magnetic polystyrene nanoparticles | Chemical immobilization via NHS/EDC method | Poly(lactide-co-glycolic acid) | Anti-PSMA antibody and paclitaxel | 57 |
| Hydrophobic nanoparticles | Block copolymerization | Glutamate | Cholesterol | 53 |
| pH- and photo-responsive nanoparticles | Single step sonication causing self-assembly of components | Selenium and dimethyl amiophenyl-rubyrin | Folic acid | 54 |
5. Stimuli-sensitive polymeric nanocarriers
Stimuli-sensitive materials can alter their physicochemical properties or structural conformations under external or internal stimuli. Stimuli-sensitive cargo delivery materials have opened a promising door to a new generation of smart delivery systems for delivering and releasing cargoes at the desired time and place. Chemical (e.g., pH), physical (e.g., temperature, light, ultrasound) and biological (e.g. enzymes) stimuli have been utilized for the design of stimuli-sensitive cargo delivery systems.62 In particular, the therapeutic benefit of a stimulus responsive material can be tremendous when the stimulus is unique to the disease pathology and the material specifically responds to the pathological “trigger”.635.1. Temperature-triggered delivery
Temperature is extensively used in stimuli-sensitive delivery because the abnormal temperatures at disease sites in the body represent a unique pathological stimulus. For instance, a tumour environment is often 1–2 °C warmer than normal tissues.64 Thermosensitive polymers have been used to develop temperature-triggered delivery systems. In general, thermo-sensitive polymers, which are used in this field exhibit a low critical solution temperature (LCST) around 38–39 °C.65,66 The thermo-sensitive polymers exhibit a hydrophilic nature in normal tissues (37 °C), but alter to hydrophobic and collapse in the pathological tumour environments (38–39 °C). This structural switching with environmental temperature change can be employed to fabricate smart nanocarriers for controllable release profiles. The poly(N-isopropylacrylamide) (NIPAM) and its copolymeric micelles with different structures (e.g., diblock, triblock, graft, branched polymers) are the most widely used polymers for thermo-responsive nanocarriers.67–69 Gan et al. prepared a series of core–shell structured PCL-b-PEO-b-PNIPAAm triblock copolymers with changing PNIPAM block lengths to investigate the performance of thermo-triggered DOX release of nanocarriers at two different incubation temperatures.70 They observed that both the PNIPAAm chain length and temperature have a great influence on the release of DOX. As seen in Fig. 6c, both polymeric particles showed a faster release rate of DOX at 42 °C (above LCST) than at 25 °C (below LCST), indicating the acceleration functions of nanoparticle response to thermo-sensitivity for drug release. They also observed that the polymeric nanocarriers of triblock copolymers with longer PNIPAAm block chains showed a slower rate of drug release than those with shorter PNIPAAm block chains with no dependence on the temperature.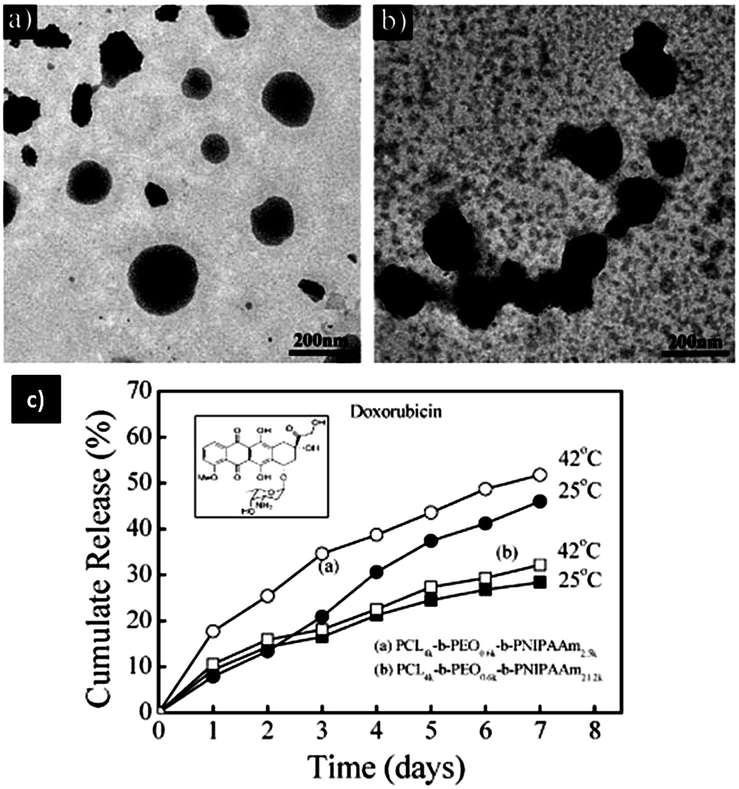 | ||
| Fig. 6 (a) TEM images of triblock copolymer nanoparticles at 25 °C, and (b) 45 °C, (c) release of DOX from nanoparticles with different PNIPAAm block length at 42 °C (>LCST) and 25 °C (<LCST). (Inset shows the molecular structure of DOX.) Adapted from ref. 70 with permission from Wiley. | ||
Sun et al. exhibited the temperature-triggered DOX release from liposomes coated with p(NIPAM-co-acrylamide) (LCST 40 °C) and PEG. They observed that the DOX release from modified liposomes was very slow below LCST. On the other hand the release of DOX from the PNIPAM-AAM/PEG modified liposomes was enhanced around the LCST of the polymer. Moreover, the stability of the PNIPAM-AAM/PEG coated liposomes was comparable with polymeric unmodified liposomes in the presence of serum because the PEG chains prevented protein adsorption.71
Polymeric nano sized micelles were also used as carriers for low-molecular-weight drugs, genes and imaging agents. Rijcken et al. synthesized core-crosslinked (CCL) biodegradable thermo-sensitive micelles based on mPEG5000 and N-(2-hydroxyethyl)methacryl-amide)-oligolactates (mPEG-b-p(HEMAm-Lacn) and investigated the physical properties of nano-micelles in vivo. They reported that the core-cross-linked micelles showed an excellent physical stability and a circulation profile compared to non-cross-linked micelles. More than 50% of the micelles still resided in the circulation 6 h post-injection, and an increased tumour accumulation was observed.72
Paasonen et al. prepared the temperature-sensitive and biodegradable poly(N-(2-hydroxypropyl)methacrylamide mono/dilactate) (pHPMA mono/dilactate) coated liposomes with a cloud point of 42 °C.73 They modified these biodegradable liposomes with cholesterol anchor to obtain the incorporation in the liposomal bilayers, and the modified liposomes were able to mediate temperature-triggered liposome aggregation and contents release. They observed that whereas the size of the uncoated liposomes remained stable upon raising the temperature from 25 to 46 °C, polymer-coated liposomes aggregated around 43 °C. In addition, the uncoated liposomes loaded with calcein hardly showed any leakage of the fluorescent marker when heated to 46 °C. On the other hand, polymer coated liposomes showed a high degree of temperature-triggered calcein release above the cloud point of the polymer.
The rapid entrapment of nanoparticles by RES is a major hindrance for in vivo systems. In this case PEGylation or other similar protective coatings create a steric barrier to prevent liposome aggregation. Moreover, the half-life of the liposomes in the bloodstream is extended.74,75 On the other hand, to enhance the temperature-triggered release from polymeric carriers, local heating of the tumour with an external heating device can subsequently trigger the drug release because of the aggregation of the thermosensitive and stable polymer carriers, followed by permeabilization of the liposomal membrane.73,76 Hyperthermia can increase tumour vessel pore size and thus increases tumour liposome extravasation. Then, hyperthermia can trigger drug release from liposomes in the tumour vessel and finally tumour cells can be directly killed. Kong et al. investigated the effects of temperatures in the range of 34–42 °C and hyperthermia treatment conditions on the extravasation of nanoparticles (100 nm liposomes) from tumour microvasculature in a human tumour (SKOV-3 ovarian carcinoma) xenograft grown in athymic nude mouse window chambers. They demonstrated almost 2–4-fold increase in uptake of (non-thermally sensitive) liposomes in heated tumours at 42 °C as opposed to non-heated tumours at 37 °C (Fig. 7a–e).77
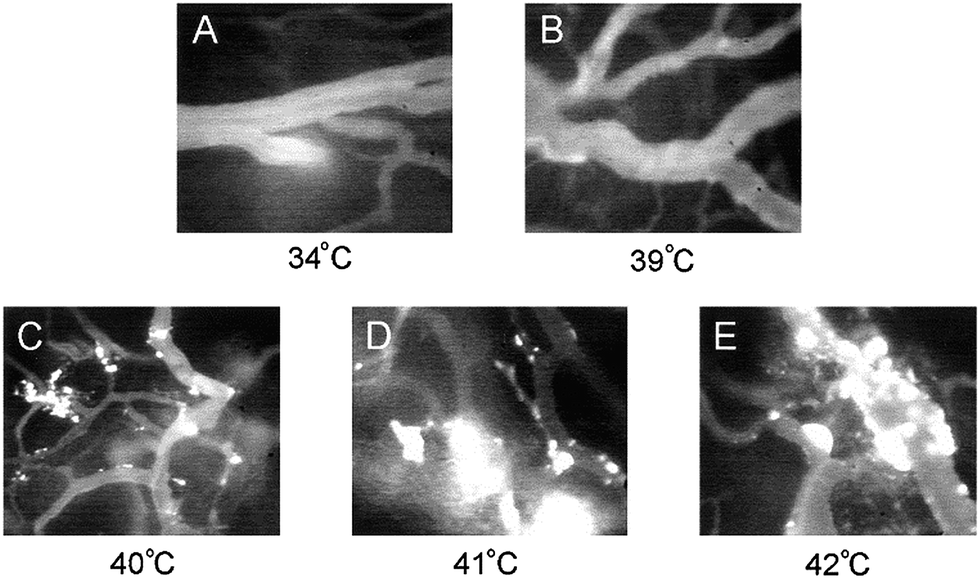 | ||
| Fig. 7 Extravasation of nanoparticles from tumour vessels at 60 min after injection at different temperatures; (a) 34 °C, (b) 39 °C, (c) 40 °C, (d) 41 °C and (e) 42 °C. Reprinted from ref. 77 with permission from AACR Publications. | ||
5.2. pH-triggered delivery
The intracellular pH of cells within healthy tissues and tumours is similar, but tumours exhibit a lower extracellular pH than normal tissues.56 The pH of blood and normal tissues is 7.4, but the extracellular pH in tumour tissues is about 6.8 because of production of lactic acid and other acids under hypoxic conditions.66 Moreover, endosomal pH may range from 4.5 to 6.5.78 The differences in pH between normal tissue and cancer tissues create an opportunity to design pH-sensitive drug delivery systems that can target tumours and release loaded drugs at the tumour site.77Recently, Ke et al. developed hollow microspheres (HMs) of poly(D,L-lactic-co-glycolic acid) (PLGA) containing sodium bicarbonate (NaHCO3) for the release of doxorubicin (DOX) in response to the acidic pH in endocytic organelles.79 In this system the crucial component is sodium bicarbonate (NaHCO3). Ke and co-workers incorporated NaHCO3 together with DOX into HMs by the use of a double-emulsion method. They observed that the HMs reached the lysosomal compartments and NaHCO3 reacted with the acid to quickly generate CO2 bubbles at near pH 5.0, which caused the microsphere wall to burst and thereby swiftly released DOX (Fig. 8).63,79 Guo et al. prepared folic acid modified-DOX-conjugated poly(ethylene glycol)–poly(ε-caprolactone) carriers for a pH-triggered drug release.80 The anticancer drug, DOX, is chemically conjugated to the polymer backbone via pH-responsive hydrazine linkers (FA-PECL-hyd-DOX). To compare the pH-triggered release performance of FA-PECL-hyd-DOX, they also synthesized carbamate-conjugated micelles (FA-PECL-cbm-DOX). They showed that the pH-sensitive FA-functionalized DOX-conjugated micelles presented considerably better efficiency of cellular uptake and higher cytotoxicity to tumour cells. In vivo pharmacokinetics and biodistribution studies indicated that.
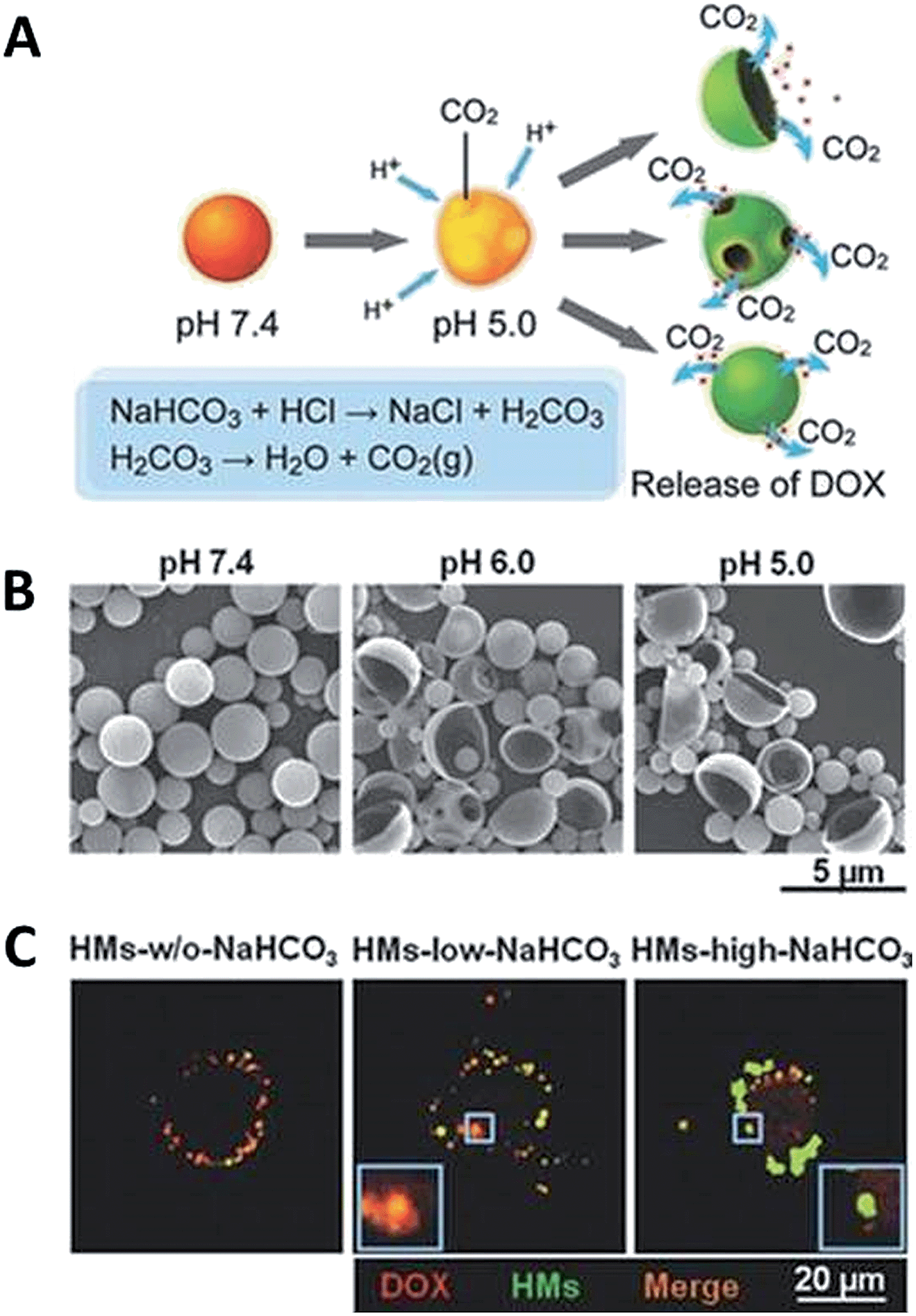 | ||
| Fig. 8 (a) Schematic illustration of the pH-responsive drug release mechanism of PLGA nanoparticles containing DOX and NaHCO3, (b) SEM images of nanoparticles after incubation in different pH, (c) CLSM images of the intracellular release of DOX from PLGA nanoparticles. Reprinted from ref. 63 with permission from Wiley. | ||
FA-PECL-hyd-DOX micelles significantly prolonged the blood circulation time of the drug and the enriched drug into the tumours rather than normal tissues. In vivo anti-tumour activity demonstrated that FA-PECL-hyd-DOX micelles had the highest safety to body and the best therapeutic efficacy to tumours.
Recently, Wang and co-workers reported the design of a smart pH- and reduction-dual-responsive drug delivery system based on folate-PEG coated polymeric lipid vesicles (∼50 nm) (FPPLVs) formed from amphiphilic dextran derivatives.81 PEG chains with pH-sensitive hydrazone bonds, stearyl alcohol (SA) chains with reduction-sensitive disulphide bonds, and folate were connected to the dextran main chain. FPPLVs carriers showed pH-triggered DOX release in response to acidic pH and reduction environments because of the cleavage of hydrazone bonds and disulphide bonds (Fig. 9). Moreover, Wang et al. demonstrated that the FPPLVs lost their PEG coating, as well as exposed the folate under acidic environment; thus, efficiently entering tumour cells through ligand–receptor interactions and in vitro cytotoxicity measurement of the FPPLVs carriers had pronounced anti-tumour activity to HeLa cells.
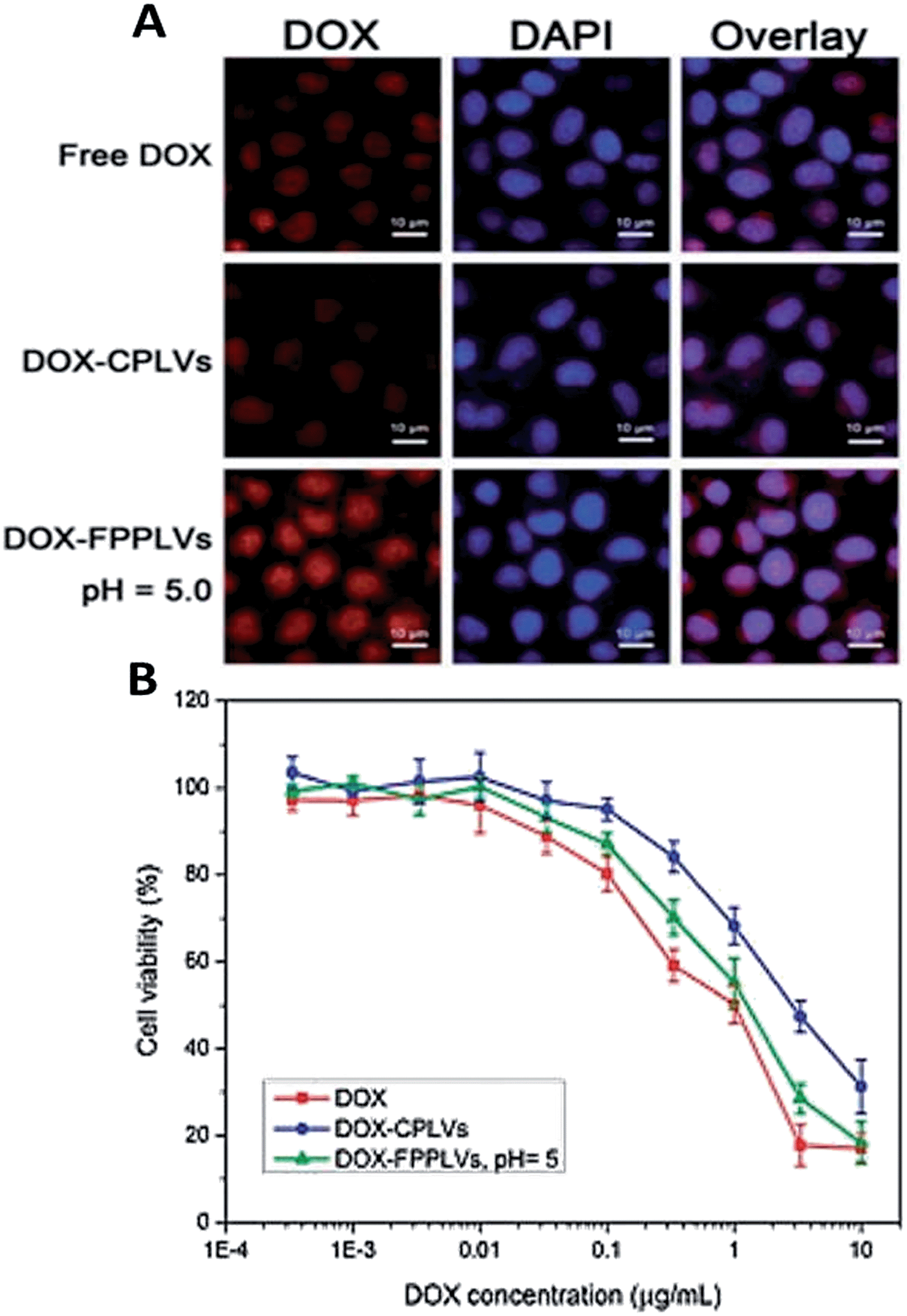 | ||
| Fig. 9 (a) Fluorescence images of HeLa cells incubated with DOX-loaded samples, (b) cell viabilities of DOX-loaded samples and free DOX as a function of DOX dosages. Reproduced from ref. 81 with permission from The Royal Society of Chemistry. | ||
5.3. Ultrasound triggered delivery
Ultrasound has a promising potential such that can be used as a trigger for cargo molecules delivery. It can penetrate deep into the body and allows for spatial and temporal control at millimetre precision.82 Ultrasound is widely used with a combination of dual-modality imaging and therapy.83,84 Basically, ultrasound can destroy the structure of nanocarriers and cause drug leakage with ultrasound-generated energy input. Thus, ultrasound can be used to control/trigger drug targeting and release from the ultrasound-sensitive nanopreparations.85We recently demonstrated the development of novel dual pH/temperature sensitive nanogel particles based on poly(vinylcaprolactam-co-2-dimethylaminoethlymethacrylate) [P(VCL-co-DMAEMA)] using surfactant free emulsion polymerization for multi-responsive release (Fig. 10a).86 The temperature/pH-dependent cumulative release and ultrasound-enhanced pulsatile release properties were investigated for RhB-loaded nanogels for long-term and one-shot delivery. The nanogels exhibit efficient delivery for both long-term and one-shot delivery systems. We observed that in pH 5.0 solution, both the DMAEMA groups of the nanogels and the RhB molecules are positively charged. Repulsive forces among the DMAEMA groups of the nanogels and the RhB molecules were stronger, and as a result, the RhB molecules are forced to be released from the nanogels. We can clearly say that P(VCL-co-DMAEMA) nanogels showed excellent tumour drug delivery character and respond to a mildly acidic environment (Fig. 10a). On the other hand, we also investigated the ultrasound-triggered release performance of the nanogels. Results showed that, for low frequency ultrasound-triggered release, the release rate strongly increases when the sample is exposed to ultrasound and it decreases when the ultrasound is stopped. As a result, ultrasound clearly caused an increase for the delivery efficiency of nanogels in our system (Fig. 10b).
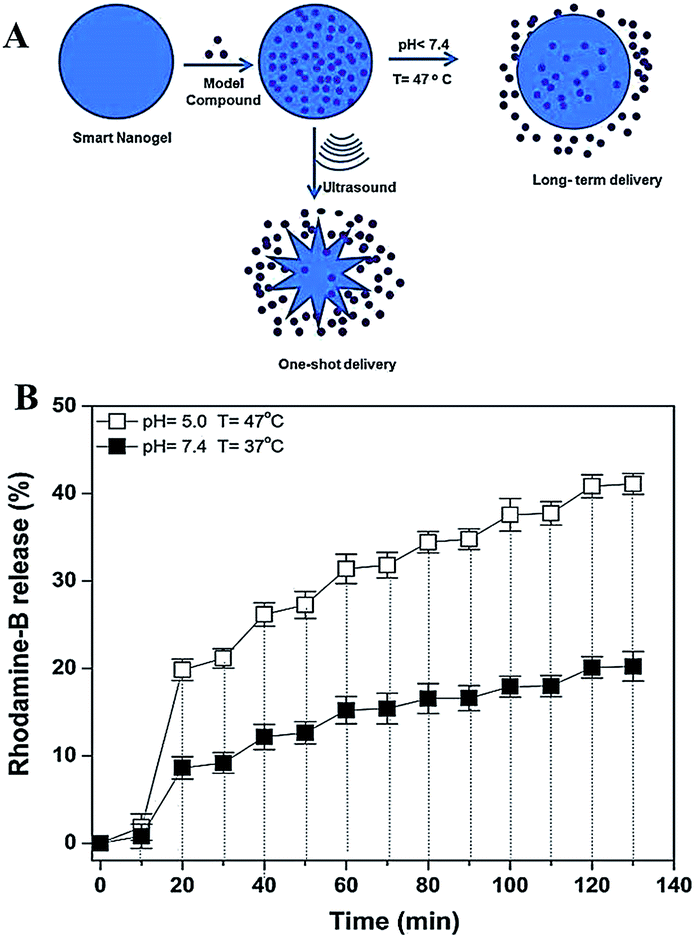 | ||
| Fig. 10 (a) The schematic drug delivery mechanism of the P(VCL-co-DMAEMA) nanogels, (b) ultrasound-triggered release of RhB from nanogels at pH 5.0, T = 47 °C and at pH 7.4, T = 37 °C. Adapted from ref. 86 with permission from Wiley. | ||
Warram et al. reported the evolution of multi-targeted micro bubbles, comparing single-, dual-, and triple-targeted motifs for ultrasound triggered efficiency. They prepared a triple-targeted microbubble coupling three antibodies against mouse aVb-integrin, P-selectin, and vascular endothelial growth factor receptor 2. They showed a 50% increase of binding affinity to mouse SVR angiosarcoma endothelial cells compared to dual-targeted microbubbles and a 40% increase in tumour image intensity compared to single- and dual-targeted microbubbles in a breast cancer-bearing mouse model.87
Phillips et al. investigated a method for gene delivery to vascular smooth muscle cells using ultrasound-triggered delivery of plasmid DNA from electrostatically coupled cationic microbubbles. Microbubbles carrying reporter plasmid DNA were acoustically ruptured in the vicinity of smooth muscle cells in vitro under a range of acoustic pressures (0 to 950 kPa) and pulse durations (0 to 100 cycles). No effect on gene transfection or cell-viability was observed from application of microbubbles, DNA, or ultrasound alone.88
5.4. Light-triggered delivery
Light is an external stimulus to trigger cargo molecule delivery. It is very attractive because of its high biocompatibility and ease of application.89–92 The tissue penetration depth is the major challenge of light-triggered delivery systems. The penetration depth is important for efficient and targeted drug release in the bulk tissue. Weissleder et al. reported that near-infrared (NIR) light with wavelengths in the range of 650 to 900 nm have an attractive optical stimulus because of the minimal attenuation by blood and soft tissues. Thus, NIR light allows for non-invasive and deep tissue penetration.93Xiao et al. developed a novel light-responsive (PnP) AZO-substituted poly(acrylic acid) template as a triggered DOX delivery derivatives system (Fig. 11).94 They showed that drug and target ligand molecules can be simultaneously loaded and unloaded onto the template by using UV irradiation. They observed that the drug loaded template cannot be taken up by normal cells because of the presence of electrostatic repulsion. They reported that the cell viability decreases to 41% as about 42% of DOX derivative is released from the template after 20 min of UV irradiation; as a control, cell viability is about 95%.
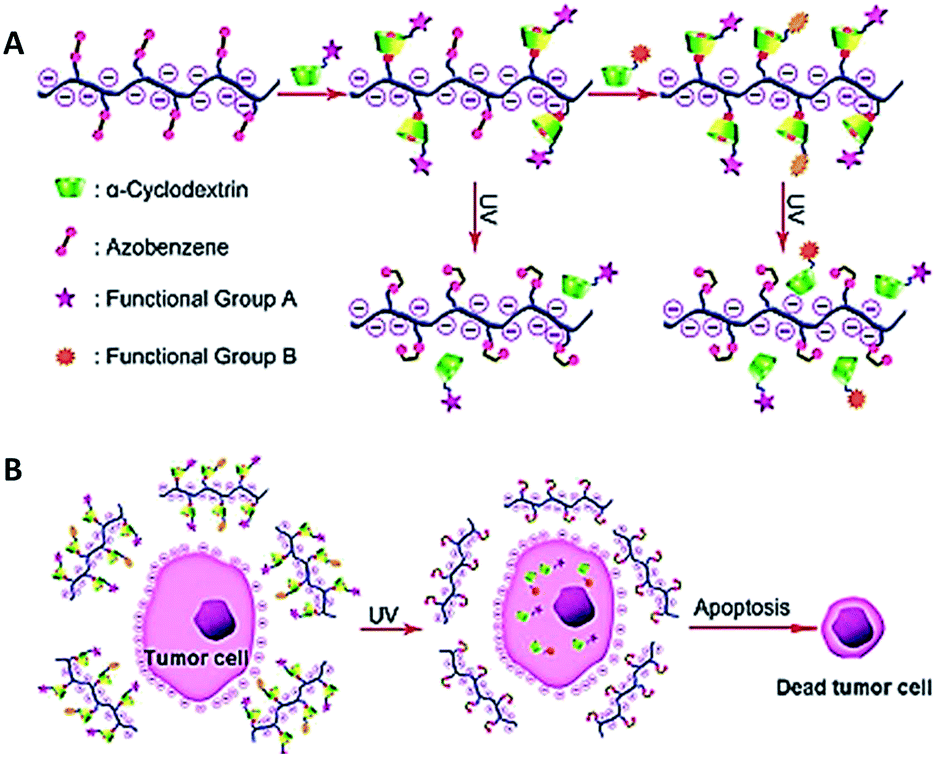 | ||
| Fig. 11 (a) Schematic illustration of the loading and release presentation of light-responsive polymers, (b) target release of DOX derivative from nanocarriers into tumor cell. Reprinted from ref. 94 with permission from Wiley. | ||
The photocleavage reaction is also used to create light-triggered polymeric nanocarriers. Many studies showed that o-nitrobenzyl (NB) and coumarin-derivative-incorporated copolymers prove an efficient mode for constructing UV/NIR-triggered systems.95,96
Li et al. fabricated amphiphilic diblock copolymer micelles for phototriggered drug release and enhancement of magnetic resonance imaging (MRI) contrast performance (Fig. 12).97 In this study Li and co-workers synthesized covalent Gd3+ labelled OEGMA-b-P(NIPAM-co-NBA-co-Gd) light-responsive diblock copolymer. They reported that after UV irradiation the hydrophobic NBA moieties transform into a hydrophilic state. In this process, upon UV irradiation, micellar cores were subjected to swelling and hydrophobic-to-hydrophilic transitions and led to ∼1.9-fold increase in the enhancement of MR imaging contrast performance. In addition, they exhibited the enhancement of DOX release rate upon UV irradiation for 12 h (∼65% DOX release) versus for 25 h (∼47% DOX release).
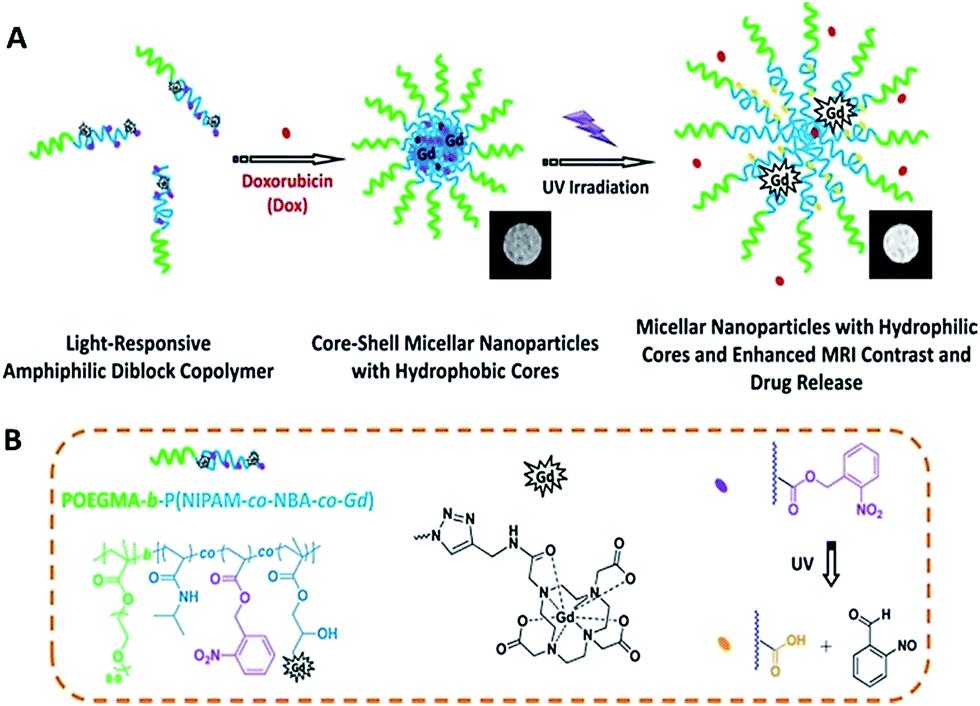 | ||
| Fig. 12 (a) Schematic illustration for the fabrication, (b) and the release mechanism of light-responsive polymeric POEGMA-b-P(NIPAM-co-NBA-co-Gd) micelles. Reprinted from ref. 97 with permission from ACS Publications. | ||
5.5. Enzyme triggered delivery
Enzymes play an important role to produce extracellular matrix (ECM)-mimetic synthetic polymeric nanocarriers for triggered smart delivery. Dong et al. reported a dual pH and enzyme responsive and tumour-specific delivery system for doxorubicin (DOX) (Fig. 13).98 They produced a negatively charged intercalation complex (polyGC-DOX) based on DOX and double-stranded oligoDNA and they mixed with C-gelatine in order to form complexes (CPX1) (Fig. 13).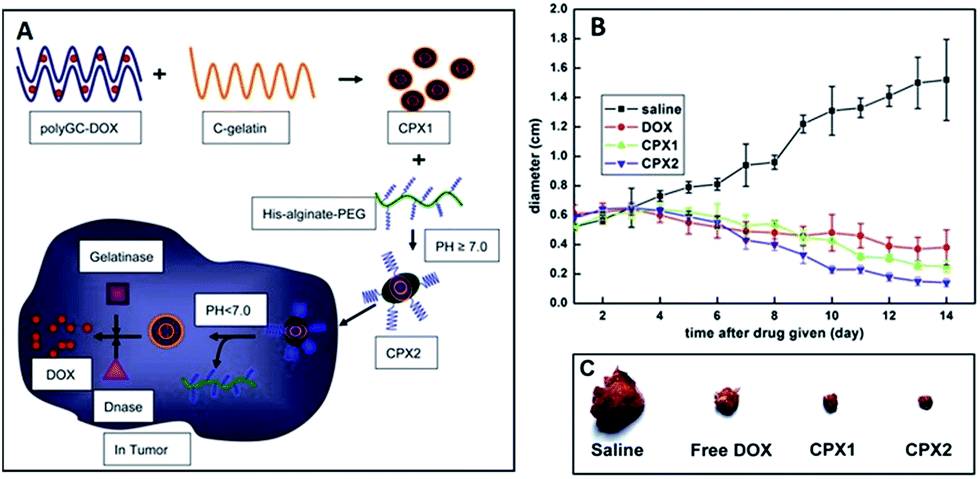 | ||
| Fig. 13 (a) Schematic illustration of the production of pH/enzyme dually responsive nanocarriers, (b) anti-tumour activity of CPX1 and CPX2 for DOX release, (c) the situations of tumours after injections of saline, free DOX, CPX1 or CPX2. Reproduced from ref. 98 with permission from Elsevier. | ||
They also combined CPX1 with a pH-sensitive PEGylated alginate to form CPX2 in order to prevent undesirable accumulation in the liver, which exhibits a relatively high concentration of gelatinase. CPX2 could be digested and release DOX under the co-digestion of gelatinase (GA) and DNase I at pH < 6.9. Thus, the nonspecific adsorption within liver was suppressed. In tumour tissue environment (pH ∼ 6.2–6.7), His-alginate-PEG dissociates from the surface of CPX2 aggregates because of electrostatic repulsion. The released CPX1 can be further co-degraded in the presence of gelatinase and deoxyribonuclease (DNase), causing the effective release of DOX (Fig. 13a). They reported that CPX2 complex increased the accumulation of DOX in tumour, reduced its deposition in heart and could specifically release DOX in tumour sites, which resulted in enhanced anti-cancer activity and decreased cardiotoxicity of DOX. They also investigated the efficiency of CPX2 complex in an animal model of implanted tumour, the CPX2 complex exhibited high effectiveness in preventing the growth of the tumours compared to free DOX as seen in Fig. 13b and c.
The changes in the composition of local enzymes such as matrix metalloproteinases (MMPs), which have been considered as biomarkers for diagnostics and prognostics in many types and stages of cancer, also provide an opportunity for delivery of drug molecules and imaging agents to pathological sites via an enzyme-triggered mechanism.99 Zhu et al. reported the design of a novel multifunctional nanocarrier as seen in Fig. 14, which responds to the overexpressed extracellular matrix metalloprotease 2 (MMP2) to enhance tumour targetability and internalization.100 They synthesized a surface-functionalized liposomal nanocarrier conjugating two functional polyethylene glycol (PEG)–lipids. Torchilin and co-workers modified the functionalized liposome with the tumour cell-specific anti-nucleosome monoclonal antibody (mAb2C5). Their studies exhibited that the produced nanocarrier accumulated in tumours and specifically targeted cancer cells. It responded to the up-regulated extracellular MMP2 in tumours, and provided enhanced cellular internalization via a cell penetration function (TATp).
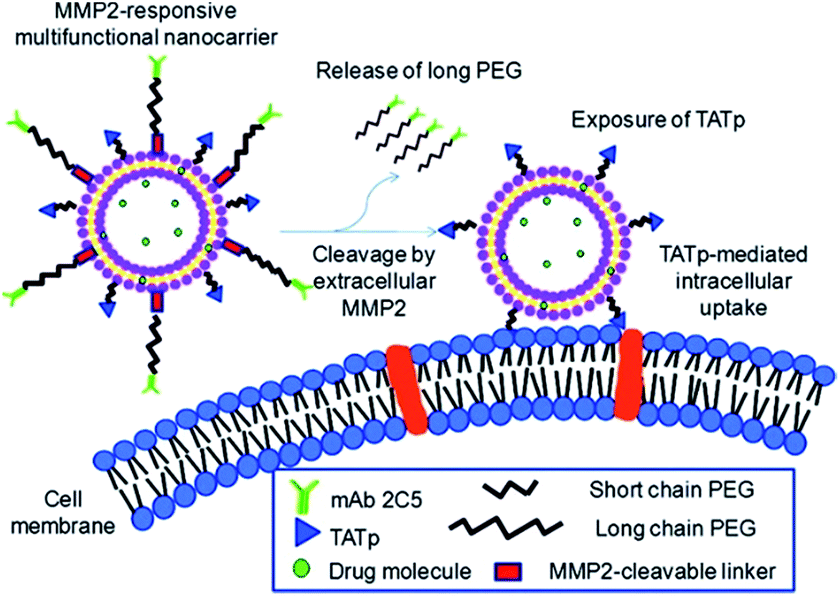 | ||
| Fig. 14 Enhanced tumour targeting using a MMP2-responsive liposomal multifunctional nanocarrier. Reprinted from ref. 100 with permission from ACS Publications. | ||
6. Bioimaging
One of the most important and extensive uses of polymers in nanomedicine is related to bioimaging, and especially in vivo applications of polymer-based bioimaging are quite prominent. The most important prerequisite of bioimaging is frequently labelling of tissues, cells, or cellular markers by various methods, among which fluorescence, radioactivity, and magnetic resonance are well established and thoroughly utilized. Fluorescent labelling of biological targets, by organic fluorophores for instance, is among the most utilized techniques for imaging, in cellular studies particularly, while widely used fluorophores possess serious drawbacks preventing their use in in vivo studies, such as low chemical stability, high toxicity, inadequate signal-to-noise ratio, and bleaching.101 Quantum dots (QDs) could be considered as a feasible alternative to fluorophore molecules; however, their high toxicity, since they frequently include heavy metals that are toxic for biological systems, is also a serious consideration.101 On the other hand, either magnetic resonance102 or radioactive imaging103 techniques require imaging agents that have been conjugated to carriers, which are mostly polymeric, in order to obtain efficient biodistribution and retention of their functional moieties. Rational design and production of polymer-based imaging systems have the possibility to provide convincing solutions to most of the aforementioned issues.Polymers in bioimaging could be used either as the imaging functionality or for enhancing any previously used imaging agent in terms of solubility, biocompatibility, efficiency etc. In any case, it is obvious that polymer-based imaging systems have considerable superiorities compared to their fluorescent, radioactive or magnetic counterparts. Particularly, in vivo imaging considerably benefits from the flexibility provided by polymer nanostructures in terms of biocompatibility and resilience. Multifunctional structures including theranostic agents could also be accomplished with polymer-based imaging systems, which is another serious advantage of using polymers for in vivo applications. For clarity, polymer-based in vivo imaging applications could be divided into two sections, depending on whether the polymers are being utilized for imaging or functionalization, where in vivo biocompatibility for all the applications that will be described are already granted. Therefore, we will focus in this section primarily on the effect of the polymers on the efficiency of in vivo imaging compared to generic imaging procedures.
6.1. All polymer fluorescence-based imaging systems
As previously described, conventional methods for fluorescent imaging have reached their natural limits, especially for in vivo applications.101 Polymer-based fluorescent probes have a significant superiority in fluorescence characteristics and biocompatibility compared to their fluorophores and QD counterparts.104 Conjugated or conducting polymers are prominent candidates for fluorescent imaging. On the other hand, their physical and chemical characteristics, such as solubility and emission wavelength need to be tuned carefully. In addition, targeting to a specific location within the body is another critical issue. One last concern is the emission wavelength, where near-infrared range is the most convenient, regarding its low absorption in a biological environment. Below, some recent in vivo applications related to the use of polymers as fluorescent probes will be summarized. For a detailed introduction and a more chemical perspective, besides critical in vitro applications, there are some recent reviews that could be referred.104–106One pioneering the application of polymers as nanoparticles for in vivo imaging is the work of Kim et al., describing the use of cyano substituted derivatives of poly(p-phenylenevinylene) (CN-PPVs) for sentinel lymph node (SLN) mapping.107 They successfully formed nanoparticles with tuneable ultrabright fluorescence by polymerization within commercially available Tween 80 micelles. These nanoparticles were demonstrated to accumulate within SLN by direct lymphatic drainage, without any specific targeting in a few minutes, and remain there for days. The SLN uptake of these nanoparticles was estimated to be 24% of the injected dose (Fig. 15).
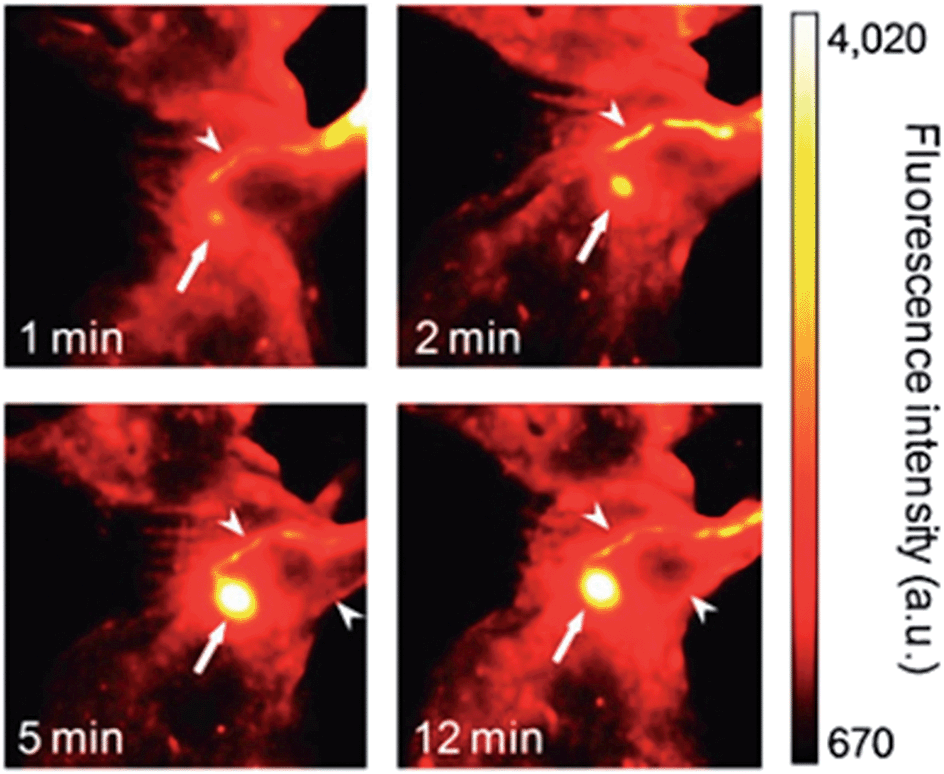 | ||
| Fig. 15 Imaging time-points of a mouse after injection of NIR-fluorescent polymer nanoparticles. Adapted from ref. 107 with permission from The Royal Society of Chemistry. | ||
Conjugated polymers could also be administrated as nanoparticles, or polymer dots (Pdots) by co-condensation of polymers with different characteristics. Wu et al. demonstrated a successful example of polymer co-condensation by combining a visible-light-harvesting polymer (PFBT) as the donor, an efficient deep-red emitting polymer (PF-DBT5) as the acceptor, and an amphiphilic polymer, poly(styrene-co-maleic anhydride) (PSMA), for the generation of surface carboxyl groups for further functionalization with chlorotoxin (CTX) that has a high affinity for tumours of neuroectodermal origin.108 The fluorescence intensity of these Pdots was measured as 15 times higher than the QDs at the same wavelength. They demonstrated in vivo imaging of brain tumours in mice, while labelling was demonstrated to occur only in mice that had the tumour, showing the efficiency and importance of targeting with CTX.
One critical issue regarding the use of organic fluorophores as ultrabright nanoparticles for imaging is aggregation caused by quenching; i.e., the loss of fluorescence because of effective intermolecular π–π stacking in the aggregates. Zhao et al. proposed a solution for this problem and demonstrated aggregate induced emission (AIE) by effectively inserting a spacer molecule, tetraphenylethene (TPE), between a pair of fluorescent dyes 3,4:9,10-tetracarboxylic perylene bisimide (PBI), and combining it with a mixture of 1,2-distearoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) and DSPE-PEG5000-folate, forming a novel conjugated polymer nanoparticle emitting fluorescence, which also red shifted, from 600 to 850 nm.109 Folate receptors are significantly expressed in H22 hepatic cancer cells inoculated to mice; therefore, these nanoparticles were shown to accumulate in the cancer tissue more significantly compared to the nanoparticles accumulated through enhanced EPR, without folate targeting, by in vivo imaging. Although they did not provide quantitative data regarding the amount of accumulation enhancement with targeting, the image they provided in the article qualitatively demonstrates an increased amount of fluorescence from the tumour (Fig. 16).
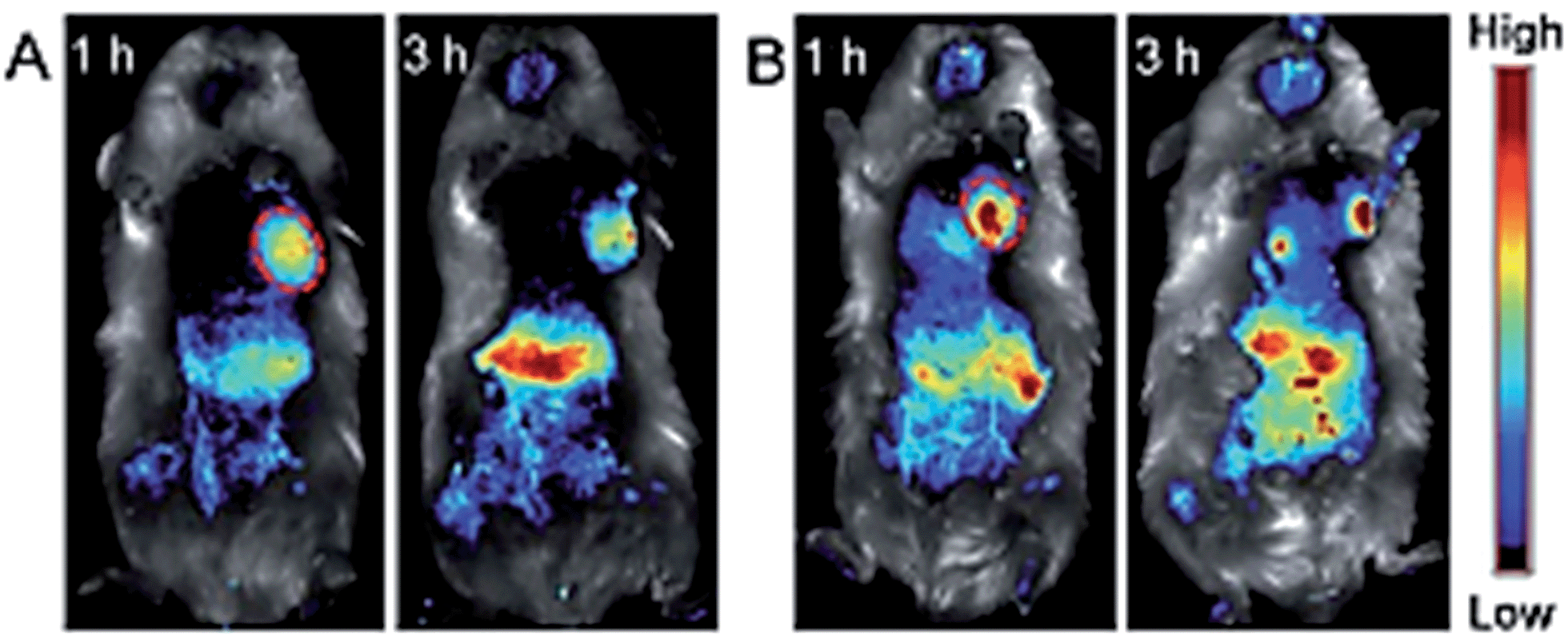 | ||
| Fig. 16 In vivo fluorescent imaging of H22 tumour-bearing mice after intravenous injection of (a) non-targeted and (b) targeted nanoparticles. Adapted from ref. 109 with permission from The Royal Society of Chemistry. | ||
Recently, the same research group has applied a similar strategy to different molecules to obtain AIE. This time they used a conjugated polymer donor (poly[9,9-bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)fluorenyldivinylene]; PFV) and a fluorogen acceptor (2-(2,6-bis((E)-4-(phenyl(4′-(1,2,2-triphenylvinyl)-[1,1′-biphenyl]-4-yl)amino)styryl)-4H-pyran-4-ylidene)malononitrile; TPE-TPA-DCM) with aggregation-induced emission characteristics using biocompatible bovine serum albumin (BSA) as the encapsulation matrix.110 After a final functionalization with arginine-glycine-aspartic acid (RGD) peptide to yield fluorescent probes for the recognition of cancer cells overexpressing integrin receptors, they observed a two-fold increase in the fluorescence obtained after RGD functionalization.
Fluorescence resonance energy transfer (FRET), or bioluminescence energy transfer (BRET) are frequently used modalities for manipulating the fluorescence characteristics of fluorophores. Especially, BRET is quite promising regarding in vivo applications, eliminating the need for the external optical excitation of the fluorophore. Xiong et al. utilized BRET for in vivo imaging of lymphatic networks and vasculature of xenografted tumours in living mice with a high tumour-to-background ratio (>100). The formed nanoparticles possessed a BRET-FRET relay capability, where chemiluminescence energy is first transferred to the polymer matrix, then to a near infrared (NIR) fluorophore.111 The most important advantage of this novel technique is that external optical stimulation is not necessary for imaging, while coelenterazine should be supplied by injection prior to imaging in order to initiate the chemiluminescence.
6.2. Polymer facilitated imaging systems
Polymers are frequently being used for enhancing particular characteristics of in vivo imaging agents other than polymers by encapsulation by various means. Some of the most important characteristics accomplished by polymer encapsulation are biocompatibility, targeted delivery, improved in vivo stability, and multifunctionality.112 Polymers are used as nanocarriers for QDs, magnetic resonant nanoparticles, and radioactive materials. The possibility of forming multifunctional in vivo imaging agents, especially for theranostics, is greatly enhanced within polymeric nanocarriers.Quantum dots are among the most studied imaging agents because of their superior characteristics compared to conventional fluorescent fluorophores, such as their ultimate fluorescence stability and excellent tenability of their fluorescence emission. On the other hand, they have serious biocompatibility deficiencies because of heavy metals constituting most of the QDs. Daou et al. investigated the effect of PEG modification of the QDs, particularly in terms of the chain length of the PEG molecules.113 QDs were observed to be deposited in the liver, spleen, skin, and bone marrow, in a PEG molecular weight-dependent manner. Especially, liver uptake of QDs was observed to reduce with PEG molecular weight greater than 12000 Da, which is expected to result in a longer blood circulation time. However, the toxicity and effect of PEGylation on toxicity of these QDs were not assessed for this particular research. Another polymer encapsulation of QDs within a PEG-based micelle is demonstrated by Pic et al. They used PEG molecules with methyl and carboxyl functionalities to coat the QDs, and they used these modified QDs for SLN imaging.114 Modified QDs accumulated within SLNs in a few minutes, and remained there for 10 days, as demonstrated by in vivo fluorescence imaging. Although their QDs were successful in imaging, they observed some toxic effects of QDs in SLNs, meaning that further research should be done for a safer use of QDs in medical applications. Moreover, these QDs could not be observed to be cleared from the body with urine, as analysed by inductively coupled plasma-mass spectroscopy (ICP-MS), which is another serious problem regarding the use of QDs for in vivo imaging.
Magnetic resonance imaging (MRI) is another non-invasive imaging modality, where imaging contrast agents, such as gadolinium ions (Gd3+) or iron oxide nanoparticles, are frequently utilized to enhance the imaging. Recently, polymers have been used for the administration of these contrast agents as nanocarriers for in vivo imaging. Shiraishi et al., for instance, prepared micelles of a block copolymer, PEG-b-poly(L-lysine) together with a Gd3+ chelator, DOTA, in order to form a nano-sized carrier for Gd3+.102 These micelles were shown to accumulate in tumours in 24 h by EPR effect. A similar approach was demonstrated by Lu et al. using amphiphilic block copolymer mPEG-b-PCL to form micelles around manganese-doped superparamagnetic iron oxide crystals.115 These micelles were shown to localize to the liver in a very short time (5 h) and remain there for 36 h (Fig. 17). They suggested that these nanoparticles could find applications in imaging lesions or tumours on the liver.
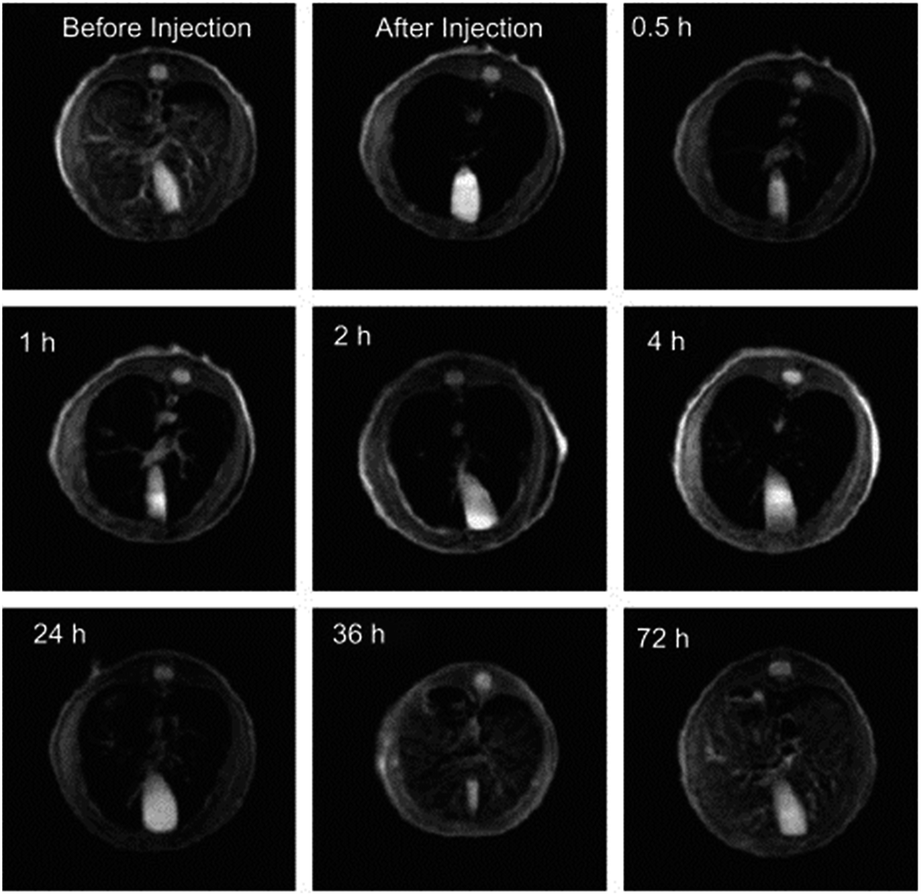 | ||
| Fig. 17 Polymer micelle encapsulated manganese-doped superparamagnetic iron oxide crystals as MRI contrast agents with respect to time. Adapted from ref. 115 with permission from Elsevier. | ||
One other broad method for non-invasive imaging is the use of radioactivity. The emissions from radioactive elements or molecules could be detected for the imaging of especially tumours. Devaraj et al. conjugated dextran-coated nanoparticles with a radioactive 18F containing molecule via click chemistry, and utilized these nanoparticles in positron emission tomography (PET) imaging.116 They measured a vascular half-life of 5.8 h for these nanoparticles, and observed accumulation within macrophages of liver, spleen, and phagocytic cells of other lymphatic organs. They also used a MRI contrast agent, while forming their nanostructure to compare the efficiency of their PET imaging method in terms of signal intensity with MIR. In addition, a NIR emitting fluorophore was used as a control during purification. They suggest that under their experimental conditions, PET is 200 times more sensitive than MRI. Herth et al. also used 18F for PET, synthesizing the macromolecules of biocompatible N-(2-hydroxypropyl)-methacrylamide (HPMA).117 They investigated the distribution and clearance of these polymers from the body by PET and observed that, while an important portion (20%) interacts with blood constituents, renal clearance is also effective in the elimination of the radioactivity. In another study by Lee et al., block copolymer micelles of PEG-b-PCL were functionalized with 111In as imaging agent, and separate nanostructures with or without epidermal growth factor (EGF) were formed as a targeting agent because EGF receptors are highly expressed in breast cancer cells.103 For imaging, they measured γ-radiation from indium-111 (In-111) after administration of micelles to mice. It is interesting that they observed only a marginal increase in the accumulation of the radioactive labelled micelles inside the tumour tissue after the targeting, which they explain with variations of tumour physiology.
The most important advantage of using polymers as nanocarriers in in vivo imaging is that it is relatively easy to obtain more than one functionality with polymers. The most profound multifunctionality that could be obtained with polymer nanocarriers is convenience for theranostic applications, while combining more than one imaging modality also provides flexibility for the utilization of the nanocarriers.
Fluorescent imaging and photodynamic therapy was combined by McCarthy et al. in order to image and interfere with atherosclerosis caused by macrophages.118 They prepared nanoparticles, which tend to selectively accumulate within macrophages. These nanoparticles contained a NIR fluorescent fluorophore and a photosensitizer. They successfully demonstrated that their nanoparticles caused apoptosis specifically in macrophages, while showing no toxic effects on nearby tissues. Lee et al. also demonstrated that glycol chitosan nanoparticles could be used for theranostic purposes when chemically conjugated with a fluorescent photosensitizer chlorin e6 (Ce6).119 They especially emphasized that conjugation of the drug is more efficient compared to physical loading by hydrophobic interactions in terms of biodistribution (Fig. 18). They also demonstrated that the photosensitizer conjugated nanoparticles showed a significant therapeutic effect compared to free Ce6 or physically loaded Ce6. In another study by Tsai et al., pH dependent dissociation of micelles prepared by the combination of graft (PEG-PLA) and diblock P(VCL-co-VM)-g-PLA were used for the controlled release of another fluorescent photosensitizer, protoporphyrin IX (PPIX).120 They observed accumulation in the brain tumour in mice because of EPR effect, an increased biodistribution of photosensitizer because of pH dependent release, and a significant anti-tumour activity.
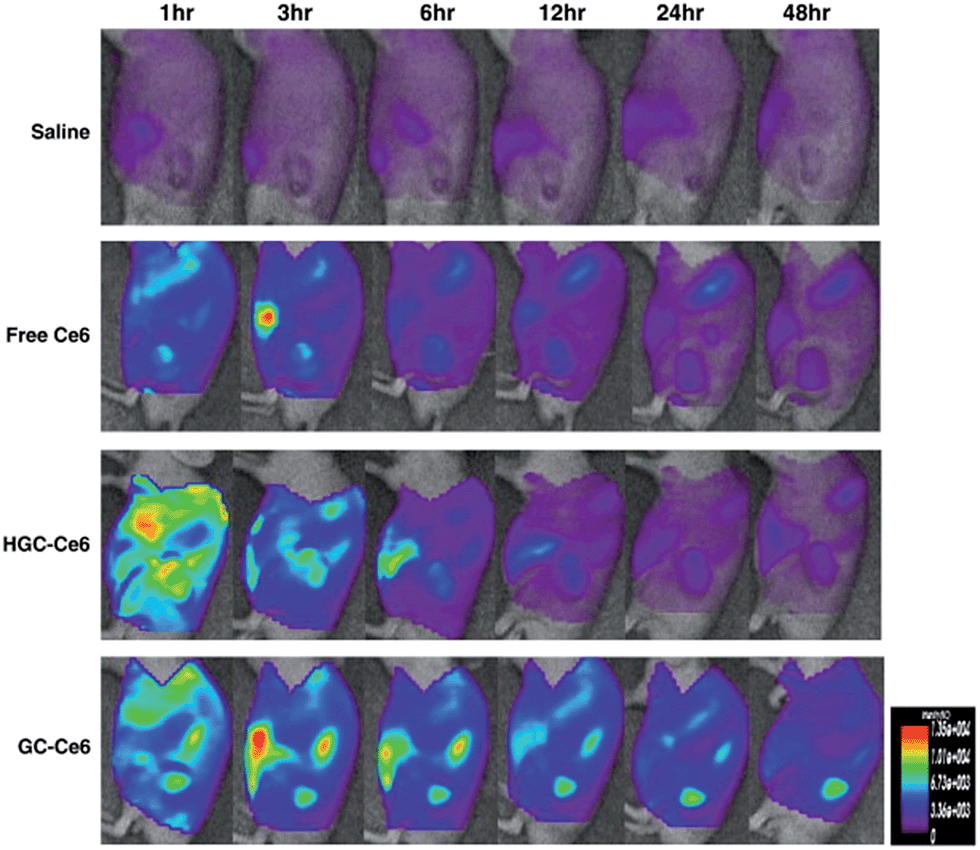 | ||
| Fig. 18 Effect of covalent conjugation of the fluorescent photosensitizer to the biodistribution with respect to time. Adapted from ref. 119 with permission from Elsevier. | ||
Polymer nanoparticles, in addition to theranostics, could also be used to combine multiple imaging modalities. Nahrendorf et al., for instance, prepared dextran-coated iron oxide nanoparticles modified with 18F for radioactive labelling, VT680 as a NIR fluorophore, and a targeting agent.121 These nanoparticles were used for simultaneous PET and optical imaging of tumour-associated macrophages in vivo. They measured a very high correlation among images obtained by fluorescence measurement and PET. The same research group also used these nanoparticles to detect the inflammation caused by macrophages in aortic aneurysms, but in that report, they did not provide any corresponding fluorescence imaging data.122 MRI contrast agents have also been used together with drugs as theranostic agents. The result demonstrated by Liu et al. is an example of such a scheme. They functionalized β-cyclodextrin-based star polymers asymmetrically with DOTA as Gd3+ chelator and doxorubicin (DOX), a frequently used anti-tumour drug, conjugated with a pH-sensitive manner.123 They also used folic acid for targeting, while pH sensitive conjugation of DOX enhances the targeted delivery approach. Despite showing successful results regarding in vitro drug efficacy and in vivo MRI, they did not provide any data related to in vivo targeting, or effect of drug on tumours in animals, since they did not perform experiments on an animal tumour model.
Results of a similar study were reported by Yang et al., which also includes PET in addition to MRI and targeted drug delivery.124 They used superparamagnetic iron oxide nanoparticles modified with PEG, then they conjugated DOX with hydrazine, a pH-sensitive linker molecule, NOTA as PET isotope (64Cu) chelator, and cyclo(Arg-Gly-Asp-D-Phe-Cys) (c(RGDfC)) for tumour targeting (Fig. 19). The report regarding those multifunctional nanoparticles also lacks in vivo targeting and drug efficiency data because a tumour model in animals was not used.
 | ||
| Fig. 19 Illustration of the multifunctional cRGD-conjugated SPIO nanocarriers for combined tumour-targeting drug delivery and PET/MR imaging. Adapted from ref. 124 with permission from Elsevier. | ||
Radioactive imaging was combined with fluorescence by Peng et al., and they also showed that their system could be used in photothermal therapy (PTT).125 They prepared micelles based on PEG-b-PCL derivatives and loaded them with IR-780 iodide as a NIR fluorophore and PTT sensitizer, then they reacted these micelles with 188Re-perrhenate (188ReO4). In vivo imaging was performed by measuring both the γ-radiation and fluorescence. PTT was applied by using light at the excitation wavelength (808 nm), and was shown to significantly diminish tumour growth.
7. Conclusion and prospects
With the development of new equipment with the ability to measure material properties in nanometer scale, researchers have focused their attention on developing materials in few nanometers with respect to clinical demands. Moreover, the remarkable increment in cancer prevalence, harmful side-effects of the drugs developed and requirement for drug delivery to the right place, at the right time and at the right dose forced the researchers to alternative approaches for cancer treatment. In this crossroad, nanoparticles, especially polymeric nanocarriers, have found a place because of their possible uses in not only diagnosis and detection of diseases, but also in the imaging of disease and monitoring the treatment. Occasionally, it is possible to develop multifunctional nanoparticles, which simultaneously act in therapy and diagnosis roles. These materials have great potential to develop new-generation theranostic agents and personalized medicine for clinical treatments. A great number of possibilities to produce and modify polymeric nanocarriers (PNCs) have formed a great expectation to challenge stemmed from the biological features, nanoparticle properties, chemical and physical properties of drugs. PNCs have great potential to develop new-generation theranostic agents and personalized medicine for clinical treatments. In addition to, the biological features, pH and temperature differences between healthy and tumour cells, biological barriers, such as skin, muscle, cellular and mucosal, should be passed for efficient drug delivery. In addition, the dispersity, stability, non-specific toxicity and half-life of the nanoparticles should be finely tuned to escape recognition by kidney and the reticuloendothelial system (RES). Although there are great expectations from the PNCs, they still face a number of challenges for biomedical uses and their further translation to clinical applications.In this review, we discuss the current challenges in respect to polymeric nanocarriers for nanomedicine and alternative ways for their solutions. In addition, we aimed at projection to illuminate situations expected in near future. In this context, polymeric nanocarriers should have the ability to access the cells targeted, while passing the biological barriers and while escaping from the RES to protect their cargo molecules from biological degradation118,126 and reducing harmful side-effects in healthy cells and tissues. As mentioned before, first the polymeric nanocarriers should escape from the RES to improve their circulation time, which is the main parameter to achieve efficient delivery of cargo molecules to target. For this aim, in addition to surface functionality, the significant parameters, i.e. shape, size and surface charge of polymeric nanocarriers, should be tuned. Herein, different materials have been used to develop effective nanosized carriers including micelles, liposomes, dendrimers and biodegradable and biocompatible polymeric nanoparticles. Moreover, the effect of the spherical,14 branch,15,16 and worm-like21 structures on circulation time, penetration, biodistribution and effective target have been discussed in literature. To improve the hydrophilicity and biocompatibility of nanocarriers, some chemical modification approaches like PEGylation have been used and its effect on the biodistribution, circulation time, carriers architecture, also drug loading capacity, drug release profile, and drug/membrane interactions.18 Recently, it has been reported that cleavable PEG ligands were more efficient in that they could enhance cellular uptake of nanocarriers.24 To adjust the surface charge, negatively/positively charged ligands have been attached on the nanocarrier surfaces and their effects on nanocarrier efficiencies have been compared.23,25 Moreover, charged distribution from center to surface has been discussed by comparing the effects of phosphonium and ammonium groups on the toxicity and transfection properties of gene delivery systems.23 The immobilization of multifunctional agents and active targeting ligands, such as antibodies, peptides, and aptamers119,127 onto nanocarriers, is one of the key steps in adjusting their features. The functionalization of nanocarriers could be performed through two different ways: starting with modified monomers and/or modifying them after synthesis to obtain efficient drug loading capacity and releasing profile. Cytotoxicity is another significant parameter being discussed to tune and reduce harmful side-effects of the therapeutic agent. Flow cytometry, microscopies including confocal, optical, fluorescent, scanning electron and transmission electron, cell viability and cell cycle analyses, microplate/ELISA readers and MTT assay have been applied to discuss the efficiency of nanoparticles developed.38–48 One of the challenges in respect to cancer chemotherapy is the dose of drug used.49 Therefore, the drug loading method and capacity should be chosen while considering its releasing profile. Physical adsorption,49 dialysis,41,42 encapsulation,44 oil-in-water single emulsion,43,46 single step sonication,47,48 core–shell entrapment,40,50 and chemical immobilization51,52 methods have been developed for adjusting the drug loading capacity while reducing harmful side-effects. In order to reduce harmful side-effects of anticancer drugs against not only targeted tumour cells, but also tissues in the delivery pathway and healthy mammalian cells, it is possible to attach specific (bio)ligands, such as folic acid,38,47 lactide and glycolic acid,39,53 antibody against fibroblast activation protein,51 beta-amino acids,40 leucine and ethylene,53 acetate/pyrrolidone42, cell penetration peptide (Cys-Tzt),54 1,4-butanediol as substrate for lipases,43 polyethylene glycol,44 tocopheryl polyethylene glycol succinate,45 anti-PSMA antibody,50 cholesterol,46 have been immobilized onto nanocarrier surfaces. As mentioned before, starting with specific functional monomers is one of the efficient ways to synthesize smart polymeric nanocarriers, which could alter their physicochemical properties or structural conformations in respect to external and/or internal stimuli. In this manner, they deliver and release their cargo molecules at the desired time and region according to chemical (e.g., pH),56,59,70–72 physical (e.g., temperature, light, ultrasound)58–66,75–79,82–87 and biological (e.g. enzymes)91–93 stimulus.
Regarding the imaging applications, one of the main challenges is the simultaneous detection of multiple targets. In this case, multifunctionality of PNCs is the key feature to develop nanotheranostics, which could be used as contrast agent for bioimaging while monitoring the pathway of drug delivery and certain releasing drug at the desired place at the right time. For this aim, various approaches such as fluorescence,97–106 radioactivity,96,108,109 and magnetic resonance95,107 labels have been applied and thoroughly utilized. A recent work of Rong et al. is a successful example of PNCs with emission line widths of 40–55 nm, facilitated by intra-chain energy transfer among conjugated BODIPY units.128 On the other hand, some multifunctional polymeric macromolecules, which contain therapeutic agents and have imaging capabilities have been under clinical investigation for a long time.130 Seymour et al. demonstrated that DOX-conjugated and 123I-labelled HPMA macromolecules could be used for theranostic purposes, and the reported toxicity levels were tolerable, while further development of the drug efficacy was required.131 Although successful targeting and imaging could be attained by polymer-based platforms, therapeutic effect would be still the primary concern regarding their large-scale clinical applications.
To date, these results were demonstrated only in vitro, and in vivo efficiency of similar methods requires further investigation. The most important thing is that the investigations of developing PNCs for clinical uses are just experimental. There is still no work that has achieved clinical standards and requirements such as reproducible, safe, clinically effective, largely scalable, and economically acceptable.129
In this review the recent advances of the polymer-based nanocarrier systems for imaging and therapy were highlighted. We discussed the promising works in the recent literature that have great potential to transfer PNCs to industry-scale clinical applications. In conclusion, PNCs are still challenging for clinical trial with their undeniable potential. It should be noted that each reported PNC system is a crucial step to achieving the development of personalized medicine. Finally, we mention that the study about developing the most efficient polymeric nanocarrier for nanotheranostics applications would exponentially grow in respect to the demand of medicinal community. We hope this review makes a significant contribution to researchers interested in this field, while compiling and comparing the recent articles.
Acknowledgements
The authors are thankful to Tamer Dogan and Prof. Gokhan Demirel for fruitful discussion.Notes and references
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- H. Wei, R. X. Zhuo and X. Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS PubMed.
- R. Cheng, F. H. Meng, C. Deng, H. A. Klok and Z. Y. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef PubMed.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Muller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2014, 39, 43–86 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, Mol. Pharm., 2010, 7, 1899–1912 CrossRef CAS PubMed.
- T. Lammers, V. Subr, K. Ulbrich, W. E. Hennink, G. Storm and F. Kiessling, Nano Today, 2010, 5, 197–212 CrossRef CAS PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- K. Kataoka, in Controlled Drug Delivery: The Next Generation, ed. K. Park, ACS, Washington, DC, 1996 Search PubMed.
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- S. Stolnik, L. Illum and S. S. Davis, Adv. Drug Delivery Rev., 2012, 64, 290–301 CrossRef PubMed.
- S. A. Kulkarni and S. S. Feng, Pharm. Res., 2013, 30, 2512–2522 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- M. Mendez-Perez, B. Vaz, L. Garcia-Rio and M. Perez-Lorenzo, Langmuir, 2013, 29, 11251–11259 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- Y. A. Zhong, F. H. Meng, C. Deng and Z. Y. Zhong, Biomacromolecules, 2014, 15, 1955–1969 CrossRef CAS PubMed.
- M. E. Fox, F. C. Szoka and J. M. J. Frechet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS PubMed.
- O. L. P. De Jesus, H. R. Ihre, L. Gagne, J. M. J. Frechet and F. C. Szoka, Bioconjugate Chem., 2002, 13, 453–461 CrossRef PubMed.
- E. R. Gillies and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124, 14137–14146 CrossRef CAS PubMed.
- S. M. Loverde, M. L. Klein and D. E. Discher, Adv. Mater., 2012, 24, 3823–3830 CrossRef CAS PubMed.
- V. V. Shuvaev, M. A. Ilies, E. Simone, S. Zaitsev, Y. Kim, S. S. Cai, A. Mahmud, T. Dziubla, S. Muro, D. E. Discher and V. R. Muzykantov, ACS Nano, 2011, 5, 6991–6999 CrossRef CAS PubMed.
- C. Ornelas-Megiatto, P. R. Wich and J. M. J. Frechet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed.
- L. Mei, L. Fu, K. Shi, Q. Zhang, Y. Liu, J. Tang, H. Gao, Z. Zhang and Q. He, Int. J. Pharm., 2014, 468, 26–38 CrossRef CAS PubMed.
- S. W. Morton, Z. Y. Poon and P. T. Hammond, Biomaterials, 2013, 34, 5328–5335 CrossRef CAS PubMed.
- Q. Peng, S. Zhang, Q. Yang, T. Zhang, X. Q. Wei, L. Jiang, C. L. Zhang, Q. M. Chen, Z. R. Zhang and Y. F. Lin, Biomaterials, 2013, 34, 8521–8530 CrossRef CAS PubMed.
- Y. Sheng, C. S. Liu, Y. Yuan, X. Y. Tao, F. Yang, X. Q. Shan, H. J. Zhou and F. Xu, Biomaterials, 2009, 30, 2340–2348 CrossRef CAS PubMed.
- S. X. Lv, W. T. Song, Z. H. Tang, M. Q. Li, H. Y. Yu, H. Hong and X. S. Chen, Mol. Pharm., 2014, 11, 1562–1574 CrossRef CAS PubMed.
- A. Yousefi, G. Storm, R. Schiffelers and E. Mastrobattista, J. Controlled Release, 2013, 170, 209–218 CrossRef CAS PubMed.
- V. R. Devadasu, V. Bhardwaj and M. Kumar, Chem. Rev., 2013, 113, 1686–1735 CrossRef CAS PubMed.
- Y. Michinaka and S. Mitragotri, J. Controlled Release, 2011, 153, 249–254 CrossRef CAS PubMed.
- S. Wang, D. Zeng, J. Niu, H. J. Wang, L. L. Wang, Q. Li, C. Y. Li, H. Song, J. Chang and L. Y. Zhang, J. Mater. Chem. B, 2014, 2, 877–884 RSC.
- A. Orthmann, R. Zeisig, T. Koklic, M. Sentjurc, B. Wiesner, M. Lemm and I. Fichtner, J. Pharm. Sci., 2010, 99, 2423–2433 CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- Y. L. Jang, U. J. Yun, M. S. Lee, M. G. Kim, S. Son, K. Lee, S. Y. Chae, D. W. Lim, H. T. Kim, S. H. Kim and J. H. Jeong, Int. J. Pharm., 2012, 434, 488–493 CrossRef CAS PubMed.
- J. F. Liu, J. J. Liu, L. P. Chu, Y. M. Wang, Y. J. Duan, L. N. Feng, C. H. Yang, L. Wang and D. L. Kong, Int. J. Nanomed., 2011, 6, 59–69 CAS.
- M. S. Cartiera, K. M. Johnson, V. Rajendran, M. J. Caplan and W. M. Saltzman, Biomaterials, 2009, 30, 2790–2798 CrossRef CAS PubMed.
- L. Costantino, F. Gandolfi, G. Tosi, F. Rivasi, M. A. Vandelli and F. Forni, J. Controlled Release, 2005, 108, 84–96 CrossRef CAS PubMed.
- H. Y. Nam, K. Nam, H. J. Hahn, B. H. Kim, H. J. Lim, H. J. Kim, J. S. Choi and J. S. Park, Biomaterials, 2009, 30, 665–673 CrossRef CAS PubMed.
- N. Voigt, P. Henrich-Noack, S. Kockentiedt, W. Hintz, J. Tomas and B. A. Sabel, Eur. J. Pharm. Biopharm., 2014, 87, 19–29 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Linden, ACS Nano, 2009, 3, 197–206 CrossRef CAS PubMed.
- P. Kocbek, S. Kralj, M. E. Kreft and J. Kristl, Eur. J. Pharm. Sci., 2013, 50, 130–138 CrossRef CAS PubMed.
- C. Y. Zhang, Y. Q. Yang, T. X. Huang, B. Zhao, X. D. Guo, J. F. Wang and L. J. Zhang, Biomaterials, 2012, 33, 6273–6283 CrossRef CAS PubMed.
- P. F. Gu, H. Xu, B. W. Sui, J. X. Gou, L. K. Meng, F. Sun, X. J. Wang, N. Qi, Y. Zhang, H. B. He and X. Tang, Int. J. Nanomed., 2012, 7, 109–122 CrossRef CAS PubMed.
- N. Bailly, M. Thomas and B. Klumperman, Biomacromolecules, 2012, 13, 4109–4117 CAS.
- J. Liu, Z. Z. Jiang, S. M. Zhang and W. M. Saltzman, Biomaterials, 2009, 30, 5707–5719 CrossRef CAS PubMed.
- J. M. Chan, L. F. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS PubMed.
- Y. F. Tan, P. Chandrasekharan, D. Maity, C. X. Yong, K. H. Chuang, Y. Zhao, S. Wang, J. Ding and S. S. Feng, Biomaterials, 2011, 32, 2969–2978 CrossRef CAS PubMed.
- Q. Ma, B. Li, Y. Y. Yu, Y. Zhang, Y. Wu, W. Ren, Y. Zheng, J. He, Y. M. Xie, X. R. Song and G. He, Int. J. Pharm., 2013, 445, 88–92 CrossRef CAS PubMed.
- J. W. Tian, L. Ding, H. J. Xu, Z. Shen, H. X. Ju, L. Jia, L. Bao and J. S. Yu, J. Am. Chem. Soc., 2013, 135, 18850–18858 CrossRef CAS PubMed.
- C. Y. Gong, X. W. Wei, X. H. Wang, Y. J. Wang, G. Guo, Y. Q. Mao, F. Luo and Z. Y. Qian, Nanotechnology, 2010, 21, 8 Search PubMed.
- N. N. Reddy, K. Varaprasad, S. Ravindra, G. V. S. Reddy, K. M. S. Reddy, K. M. M. Reddy and K. M. Raju, Colloids Surf., A, 2011, 385, 20–27 CrossRef CAS PubMed.
- H. S. Cho, Z. Y. Dong, G. M. Pauletti, J. M. Zhang, H. Xu, H. C. Gu, L. M. Wang, R. C. Ewing, C. Huth, F. Wang and D. L. Shi, ACS Nano, 2010, 4, 5398–5404 CrossRef CAS PubMed.
- S. K. E. Messerschmidt, A. Musyanovych, M. Altvater, P. Scheurich, K. Pfizenmaier, K. Landfester and R. E. Kontermann, J. Controlled Release, 2009, 137, 69–77 CrossRef CAS PubMed.
- A. Ponta and Y. Bae, Pharm. Res., 2010, 27, 2330–2342 CrossRef CAS PubMed.
- R. H. Fang, S. Aryal, C. M. J. Hu and L. F. Zhang, Langmuir, 2010, 26, 16958–16962 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- Y. Li, G. H. Gao and D. S. Lee, Adv. Healthcare Mater., 2013, 2, 388–417 CrossRef CAS PubMed.
- Y. Wang, M. S. Shim, N. S. Levinso, H.-W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS.
- J. A. Mackay and A. Chilkoti, Int. J. Hyperther., 2008, 24, 483–495 CrossRef PubMed.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. H. Qin, Y. Geng, D. E. Discher and S. Yang, Adv. Mater., 2006, 18, 2905–2909 CrossRef CAS.
- J. Qian and F. P. Wu, Chem. Mater., 2007, 19, 5839–5841 CrossRef CAS.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197–221 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS PubMed.
- P. J. Sun, Y. Zhang, L. Q. Shi and Z. H. Gan, Macromol. Biosci., 2010, 10, 621–631 CrossRef CAS PubMed.
- H. D. Han, B. C. Shin and H. S. Choi, Eur. J. Pharm. Biopharm., 2006, 62, 110–116 CrossRef CAS PubMed.
- C. J. Rijcken, C. J. Snel, R. M. Schiffelers, C. F. van Nostrum and W. E. Hennink, Biomaterials, 2007, 28, 5581–5593 CrossRef CAS PubMed.
- L. Paasonen, B. Romberg, G. Storm, M. Yliperttula, A. Urtti and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2131–2136 CrossRef CAS PubMed.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- N. Z. Wu, D. Da, T. L. Rudoll, D. Needham, A. R. Whorton and M. W. Dewhirst, Cancer Res., 1993, 53, 3765–3770 CAS.
- A. M. Ponce, Z. Vujaskovic, F. Yuan, D. Needham and M. W. Dewhirst, Int. J. Hyperthermia, 2006, 22, 205–213 CrossRef CAS PubMed.
- G. Kong, R. D. Braun and M. W. Dewhirst, Cancer Res., 2001, 61, 3027–3032 CAS.
- J. X. Gu, W. P. Cheng, J. G. Liu, S. Y. Lo, D. Smith, X. Z. Qu and Z. Z. Yang, Biomacromolecules, 2008, 9, 255–262 CrossRef CAS PubMed.
- C. J. Ke, T. Y. Su, H. L. Chen, H. L. Liu, W. L. Chiang, P. C. Chu, Y. N. Xia and H. W. Sung, Angew. Chem., Int. Ed., 2011, 50, 8086–8089 CrossRef CAS PubMed.
- X. Guo, C. L. Shi, J. Wang, S. B. Di and S. B. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS PubMed.
- S. Wang, H. Wang, Z. Liu, L. Wang, X. Wang, L. Su and J. Chang, Nanoscale, 2014, 6, 7635–7642 RSC.
- G. A. Husseini and W. G. Pitt, J. Pharm. Sci., 2009, 98, 795–811 CrossRef CAS PubMed.
- Y. Inoue, K. Goto, T. Hayashi and M. Hayashi, Int. J. Urol., 2011, 18, 358–363 CrossRef PubMed.
- S. E. Jung, S. H. Cho, J. H. Jang and J. Y. Han, Abdom. Imag., 2011, 36, 185–195 CrossRef PubMed.
- L. Zhu and V. P. Torchilin, Integr. Biol., 2013, 5, 96–107 RSC.
- G. B. Demirel and R. von Klitzing, ChemPhysChem, 2013, 14, 2833–2840 CrossRef CAS PubMed.
- J. M. Warram, A. G. Sorace, R. Saini, H. R. Umphrey, K. R. Zinn and K. Hoyt, J. Ultrasound Med., 2011, 30, 921–931 Search PubMed.
- L. C. Phillips, A. L. Klibanov, B. R. Wamhoff and J. A. Hossack, Ultrasound Med. Biol., 2010, 36, 1470–1480 CrossRef PubMed.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- N. Nishiyama, A. Iriyama, W. D. Jang, K. Miyata, K. Itaka, Y. Inoue, H. Takahashi, Y. Yanagi, Y. Tamaki, H. Koyama and K. Kataoka, Nat. Mater., 2005, 4, 934–941 CrossRef CAS PubMed.
- A. Kocer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef CAS PubMed.
- G. H. Wu, A. Milkhailovsky, H. A. Khant, C. Fu, W. Chiu and J. A. Zasadzinski, J. Am. Chem. Soc., 2008, 130, 8175–8177 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- W. Xiao, W. H. Chen, X. D. Xu, C. Li, L. Zhang, R. X. Zhuo and X. Z. Zhang, Adv. Mater., 2011, 23, 3526–3550 CrossRef CAS PubMed.
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005–1020 CrossRef CAS PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- Y. M. Li, Y. F. Qian, T. Liu, G. Y. Zhang and S. Y. Liu, Biomacromolecules, 2012, 13, 3877–3886 CrossRef CAS PubMed.
- L. Dong, S. H. Xia, K. Wu, Z. Huang, H. A. Chen, J. N. Chen and J. F. Zhang, Biomaterials, 2010, 31, 6309–6316 CrossRef CAS PubMed.
- A. M. Mansour, J. Drevs, N. Esser, F. M. Hamada, O. A. Badary, C. Unger, I. Fichtner and F. Kratz, Cancer Res., 2003, 63, 4062–4066 CAS.
- L. Zhu, P. Kate and V. P. Torchilin, ACS Nano, 2012, 6, 3491–3498 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- K. Shiraishi, K. Kawano, T. Minowa, Y. Maitani and M. Yokoyama, J. Controlled Release, 2009, 136, 14–20 CrossRef CAS PubMed.
- H. Lee, B. Hoang, H. Fonge, R. M. Reilly and C. Allen, Pharm. Res., 2010, 27, 2343–2355 CrossRef CAS PubMed.
- C. F. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- L. H. Feng, C. L. Zhu, H. X. Yuan, L. B. Liu, F. T. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- J. W. Li, J. Liu, C. W. Wei, B. Liu, M. O'Donnell and X. H. Gao, Phys. Chem. Chem. Phys., 2013, 15, 17006–17015 RSC.
- S. Kim, C. K. Lim, J. Na, Y. D. Lee, K. Kim, K. Choi, J. F. Leary and I. C. Kwon, Chem. Commun., 2010, 46, 1617–1619 RSC.
- C. F. Wu, S. J. Hansen, Q. O. Hou, J. B. Yu, M. Zeigler, Y. H. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430–3434 CrossRef CAS PubMed.
- Q. L. Zhao, K. Li, S. J. Chen, A. J. Qin, D. Ding, S. Zhang, Y. Liu, B. Liu, J. Z. Sun and B. Tang, J. Mater. Chem., 2012, 22, 15128–15135 RSC.
- D. Ding, K. Li, W. Qin, R. Y. Zhan, Y. Hu, J. Z. Liu, B. Z. Tang and B. Liu, Adv. Healthcare Mater., 2013, 2, 500–507 CrossRef CAS PubMed.
- L. Q. Xiong, A. J. Shuhendler and J. H. Rao, Nat. Commun., 2012, 3, 1193 CrossRef PubMed.
- B. T. Luk, R. H. Fang and L. F. Zhang, Theranostics, 2012, 2, 1117–1126 CrossRef CAS PubMed.
- T. J. Daou, L. Li, P. Reiss, V. Josserand and I. Texier, Langmuir, 2009, 25, 3040–3044 CrossRef CAS PubMed.
- E. Pic, T. Pons, L. Bezdetnaya, A. Leroux, F. Guillemin, B. Dubertret and F. Marchall, Mol. Imag. Biol., 2010, 12, 394–405 CrossRef PubMed.
- J. Lu, S. L. Ma, J. Y. Sun, C. C. Xia, C. Liu, Z. Y. Wang, X. N. Zhao, F. B. Gao, Q. Y. Gong, B. Song, X. T. Shuai, H. Ai and Z. W. Gu, Biomaterials, 2009, 30, 2919–2928 CrossRef CAS PubMed.
- N. K. Devaraj, E. J. Keliher, G. M. Thurber, M. Nahrendorf and R. Weissleder, Bioconjugate Chem., 2009, 20, 397–401 CrossRef CAS PubMed.
- M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rosch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef CAS PubMed.
- J. R. McCarthy, E. Korngold, R. Weissleder and F. A. Jaffer, Small, 2010, 6, 2041–2049 CrossRef CAS PubMed.
- S. J. Lee, H. Koo, H. Jeong, M. S. Huh, Y. Choi, S. Y. Jeong, Y. Byun, K. Choi, K. Kim and I. C. Kwon, J. Controlled Release, 2011, 152, 21–29 CrossRef CAS PubMed.
- H. C. Tsai, C. H. Tsai, S. A. Y. Lin, C. R. Jhang, Y. S. Chiang and G. H. Hsiue, Biomaterials, 2012, 33, 1827–1837 CrossRef CAS PubMed.
- M. Nahrendorf, E. Keliher, B. Marinelli, P. Waterman, P. F. Feruglio, L. Fexon, M. Pivovarov, F. K. Swirski, M. J. Pittet, C. Vinegoni and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7910–7915 CrossRef PubMed.
- M. Nahrendorf, E. Keliher, B. Marinelli, F. Leuschner, C. S. Robbins, R. E. Gerszten, M. J. Pittet, F. K. Swirski and R. Weissleder, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 750–U767 CrossRef CAS PubMed.
- T. Liu, X. J. Li, Y. F. Qian, X. L. Hu and S. Y. Liu, Biomaterials, 2012, 33, 2521–2531 CrossRef CAS PubMed.
- X. Q. Yang, H. Hong, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, Y. L. Xiao, Y. A. Yang, Y. Zhang, R. Nickles, W. B. Cai, D. A. Steeber and S. Q. Gong, Biomaterials, 2011, 32, 4151–4160 CrossRef CAS PubMed.
- C. L. Peng, Y. H. Shih, P. C. Lee, T. M. H. Hsieh, T. Y. Luo and M. J. Shieh, ACS Nano, 2011, 5, 5594–5607 CrossRef CAS PubMed.
- S. Dufort, L. Sancey and J. L. Coll, Adv. Drug Delivery Rev., 2012, 64, 179–189 CrossRef CAS PubMed.
- A. Z. Wang, F. Gu, L. F. Zhang, J. M. Chan, A. Radovic-Moreno, M. R. Shaikh and O. C. Farokhzad, Expert Opin. Biol. Ther., 2008, 8, 1063–1070 CrossRef CAS PubMed.
- Y. Rong, C. F. Wu, J. B. Yu, X. J. Zhang, F. M. Ye, M. Zeigler, M. E. Gallina, I. C. Wu, Y. Zhang, Y. H. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376–384 CrossRef CAS PubMed.
- B. M. Holzapfel, J. C. Reichert, J. T. Schantz, U. Gbureck, L. Rackwitz, U. Noth, F. Jakob, M. Rudert, J. Groll and D. W. Hutmacher, Adv. Drug Delivery Rev., 2013, 65, 581–603 CrossRef CAS PubMed.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281–290 CrossRef CAS.
- L. W. Seymour, D. R. Ferry, D. J. Kerr, D. Rea, M. Whitlock, R. Poyner, C. Boivin, S. Hesslewood, C. Twelves, R. Blackie, A. Schatzlein, D. Jodrell, D. Bissett, H. Calvert, M. Lind, A. Robbins, S. Burtles, R. Duncan and J. Cassidy, Int. J. Oncol., 2009, 34, 1629–1636 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |