
Dextran-based thermo-responsive hemoglobin–polymer conjugates with oxygen-carrying capacity†
Abstract
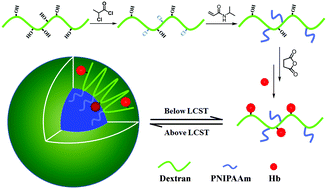
The potential toxicity towards human kidneys of hemoglobin (Hb), when used directly, has severely limited its application as a red blood cell substitute and in cancer treatments. In this work, a novel hemoglobin–polymer conjugate was prepared by a reaction between the lysine amino groups of Hb and the carboxyl groups of a copolymer, poly(N-isopropylacrylamide) grafted carboxylated dextran (HOOC-Dex-g-PNIPAAm), which was synthesized by single electron transfer living radical polymerization (SET-LRP) and post-carboxylation. The conjugate was characterized by FTIR, 1H NMR, SDS-PAGE, DLS, fluorescence spectroscopy and TEM. Results showed it had a relatively low critical micelle concentration (CMC) and could form stable spherical nanoparticles upon heating above the LCST. The redox activity and gas-binding capacity of the Hb conjugate were subsequently confirmed by UV-vis spectroscopy, indicating the retention of Hb bioactivity after conjugation. Furthermore, dextran with different numbers of PNIPAAm chains was synthesized for comparison. It revealed that at 37 °C, the temperature above the LCST, the conjugation of Hb to the copolymer Dex-g-PNIPAAm could improve the stability of Hb which increased with the number of PNIPAAm chains.
 Please wait while we load your content...
Please wait while we load your content...

