DOI:
10.1039/C4RA06313A
(Paper)
RSC Adv., 2014,
4, 41659-41665
Sterically hindered selenoether ligands: palladium(II) complexes as catalytic activators for Suzuki–Miyaura coupling†
Received
27th June 2014
, Accepted 28th August 2014
First published on 28th August 2014
Abstract
2-Hydroxy/(benzyloxy)-3,5-di-tert-butyl benzaldehyde reacts with PhSeCH2CH2NH2 resulting in a sterically hindered selenoether ligand (Schiff base) [2–HO–3,5–(C(CH3)3)2–C6H2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–(CH2)2SePh] (L1)/[2–PhCH2O–3,5–(C(CH3)3)2–C6H2–CH2–NH–(CH2)2SePh] (L2). The reactions of L1 and L2 with Na2PdCl4 in methanol and acetone–water mixture at room temperature have resulted in complexes [PdCl (L1-H)] (1) and [PdCl2(L2)] (2)], respectively. Both the complexes and their ligands have been characterized with 1H, 13C{1H} and 77Se{1H} NMR spectroscopy. The molecular structures of complexes 1 and 2 have been determined with single crystal X-ray diffraction. The Pd–Se bond lengths in 1 and 2 are 2.370(1) and 2.366(1) Å, respectively. The geometry around palladium in both the complexes is nearly square planar. Complexes 1 and 2 (0.1 mol% Pd) have been found efficient as catalysts for Suzuki–Miyaura C–C coupling reactions in the presence of K2CO3 in ethanol. The catalysis in water with complex 1 in the presence of K2CO3 was found feasible but with low conversion (up to 40%). The efficiency of 1 in carrying out the coupling is marginally better than that of 2.
N–(CH2)2SePh] (L1)/[2–PhCH2O–3,5–(C(CH3)3)2–C6H2–CH2–NH–(CH2)2SePh] (L2). The reactions of L1 and L2 with Na2PdCl4 in methanol and acetone–water mixture at room temperature have resulted in complexes [PdCl (L1-H)] (1) and [PdCl2(L2)] (2)], respectively. Both the complexes and their ligands have been characterized with 1H, 13C{1H} and 77Se{1H} NMR spectroscopy. The molecular structures of complexes 1 and 2 have been determined with single crystal X-ray diffraction. The Pd–Se bond lengths in 1 and 2 are 2.370(1) and 2.366(1) Å, respectively. The geometry around palladium in both the complexes is nearly square planar. Complexes 1 and 2 (0.1 mol% Pd) have been found efficient as catalysts for Suzuki–Miyaura C–C coupling reactions in the presence of K2CO3 in ethanol. The catalysis in water with complex 1 in the presence of K2CO3 was found feasible but with low conversion (up to 40%). The efficiency of 1 in carrying out the coupling is marginally better than that of 2.
Introduction
The strong electron-donor ability of selenium has led to the synthesis of many transition metal complexes of organoselenium ligands, which have been found promising as catalysts for various organic transformations.1 This has made organoselenium ligands currently important for designing catalysts for a variety of organic reactions. A wide range of such ligands is known e.g. selenocarbonyl,2 pincer type,3 and Schiff bases4 promising for catalyst designing. One notable feature of transition metal complexes of organoselenium ligands is that they are much less insensitive to air and moisture in comparison to those which have traditional phosphorous donors. At present palladium complexes of organoselenium ligands in terms of catalytic activity are considered not only rivals of their respective phosphorus and sulphur analogues but have been found in many cases to outperform them.3a
The steric properties of ligands significantly influence the rate and selectivity of the reactions catalyzed by their transition metal complexes. The steric bulk of a ligand can increase the stability of the catalyst in its coordinatively unsaturated intermediate form or accelerate some steps, such as reductive elimination in cross-coupling reactions which are sterically sensitive.5 Thus steric bulk of a ligand may play a role in tailoring efficiency of its complex as a catalyst for Suzuki–Miyaura cross-coupling reactions.6 Palladium complexes derived from electron-rich and sterically demanding ligands, of the type monophosphanes,7 N-heterocyclic carbenes,8 and C2-symmetric bis-hydrazone,9 have been found effective catalyst for Suzuki–Miyaura cross-coupling reactions. However no report on sterically hindered organoselenium ligands is in our knowledge inspite of several reports on activation of Suzuki–Miyaura coupling with palladium complexes and palladacycles10 of several organochalcogen donors including Se ones,4a,c–e,11 and chalcogenated carbenes,12 which have high efficiency and can be modified with ease.
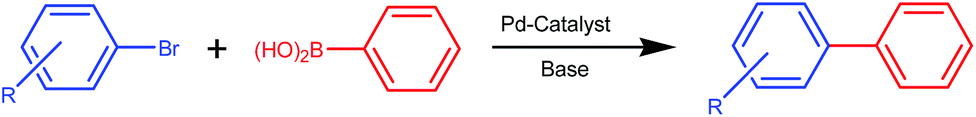
We report herein the synthesis and structural characterization of sterically hindered selenoether ligands, [2–HO–3,5–(C(CH3)3)2–C6H2–CN–(CH2)2SePh] (L1) and [2–PhCH2O–3,5–(C(CH3)3)2–C6H2–CH2–NH–(CH2)2SePh] (L2) and their palladium(II) complexes of the type, [PdCl (L1-H)] (1) and [PdCl2(L2)] (2)], respectively. Two complexes 1 and 2 have been found suitable activators for Suzuki and Miyaura C(sp2)–C(sp2) coupling reactions, which are powerful synthetic tools in organic synthesis,13,14 along with other transition metal-catalyzed carbon–carbon bond forming reactions such as Heck, Sonogashira, Hiyama, Stille and Kumada coupling reactions. Suzuki and Miyaura coupling has made a significantly higher impact than others in the laboratory and the chemical industries due to three key reasons: (i) the reaction is feasible with a wide range of substrates and many functional groups are tolerated due to mild reaction conditions. This is very helpful in the total synthesis of complex molecules including drugs. (ii) Phenylboronic acid, starting material is readily available, stable and sustainable.15 (iii) The product biaryl is a very important core component of various biologically and pharmaceutically important compounds (viz. anti-hypertensive, anti-cancer, anti-biotic, anti-inflammatory, and antifungal) and in nonlinear optical materials.16
Experimental section
Materials and methods
Diphenyl diselenide, 2-hydroxy-3,5-di-tert-butylbenzaldehyde, 2-chloroethyl amine, sodium tetrachloropalladate, phenylboronic acid, potassium carbonate, and aryl bromides were procured from Sigma-Aldrich (USA). Reagents (commercially available from local sources) were used as received without further purification. PhSe(CH2)2NH2 and 2-(benzyloxy)-3,5-di-tert-butylbenzaldehyde were prepared by a following the methods reported in the literature.17,18 The progress of every coupling reaction was monitored with NMR spectroscopy. The products of Suzuki reactions were authenticated by matching their spectroscopic data with the reported literature values. 1H, 13C{1H} and 77Se{1H} NMR spectra were recorded on a Bruker Spectrospin DPX 300 NMR spectrometer at 300.13, 75.47 and 57.24 MHz respectively. The chemical shifts are reported in ppm relative to internal standard (tetramethylsilane in case of 1H, and 13C{1H} NMR and Me2Se for 77Se{1H} NMR). Elemental analyses were carried out with a Perkin-Elmer 2400 Series II C, H, N analyzer.
X-ray diffraction data of crystals of 1 were collected on a Bruker AXS SMART-APEX diffractometer with a CCD area detector.19 Similar data of 2 were collected on an Oxford Xcalibur S diffractometer with Sapphire-3 CCD detector.20 Mo Kα radiations were used in both the cases. Both Crys Alis Pro software suite21 and SADABS22 software were used as per requirement. The structures were refined using the SHELX-97 program package and SHELXL97 (within the Win GX program package).23–26 Non-hydrogen atoms were refined anisotropically. The molecular structures were created with the Diamond program.27 Crystallographic data are given in Table 1.
Table 1 Crystallographic data and structure refinement summary for 1 and 2
| |
1 |
2·CH3CN |
| R1 = Σ||Fo| − |Fc||/Σ|Fo|. wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. S = {Σ[w(Fo2 − Fc2)2]/(n − p)2]}1/2. |
| Formula |
C23H30NOPdClSe |
C32H41N2OPdCl2Se |
| Formula weight |
557.29 |
725.93 |
| T/K |
298(2) |
298(2) |
| λ/Å |
0.71073 |
0.71073 |
| Crystal system |
Orthorhombic |
Monoclinic |
| Space group |
Pbca |
P21/c |
| a/Å |
10.9228(4) |
12.3315(5) |
| b/Å |
10.0631(4) |
10.4554(4) |
| c/Å |
43.0466(17) |
26.1643(10) |
| α/deg |
90.00 |
90.00 |
| β/deg |
90.00 |
97.856(4) |
| γ/deg |
90.00 |
90.00 |
| Vol/Å3 |
4731.5 (3) |
3341.7 (2) |
| Z |
8 |
4 |
| Dcalcd/g cm−3 |
1.565 |
1.443 |
| F(000) |
2240 |
1476 |
| θrange/deg |
2.91–25.00 |
3.06–25.00 |
| Reflections measured |
4159 |
5885 |
| Reflections used |
3702 |
5337 |
| Parameters |
259 |
359 |
| μ(Mo Kα) (cm−1) |
2.449 |
1.830 |
| R1, wR2[I > 2σ(I)]a |
0.0488, 0.0558 |
0.0357, 0.0407 |
| R1, wR2 (all data)b |
0.1014, 0.1040 |
0.0818, 0.0840 |
| GooFc |
1.302 |
1.216 |
Synthesis of L1. The selenated amine C6H5Se–(CH2)2–NH2 (0.412 g, 2.06 mmol) and 2-hydroxy-3,5-di-tert-butylbenzaldehyde (0.469 g, 2.00 mmol) were reacted in absolute ethanol (20 mL) at room temperature for 12 h. The volatiles from resulting reaction mixture were removed using rotary evaporator which resulted ligand L1 as yellow viscous liquid in 93% yield (0.776 g, 1.86 mmol). 1H NMR (CDCl3): δ (ppm) = 1.36 (s, 9H, C(CH3)3), 1.51 (s, 9H, C(CH3)3), 3.24 (t, 2H, SeCH2), 3.91 (t, 2H, NCH2), 7.12–7.58 (7H, Ar–H), 8.35 (S, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) N), 13.55 (broads, 1H, OH). 13C{1H} (CDCl3): δ (ppm) = 28.3 (SeCH2), 29.5 and 31.5 (C(CH3)3), 34.1 and 35.0 (C(CH3)3), 59.6 (NCH2), 117.7, 126.0, 127.1, 129.2, 129.6, 132.9, 136.7, 140.1, 158.0 (Ar–C) 166.9 (NCH). (77Se{1H} NMR, CDCl3): δ (ppm) = 281.1.
N), 13.55 (broads, 1H, OH). 13C{1H} (CDCl3): δ (ppm) = 28.3 (SeCH2), 29.5 and 31.5 (C(CH3)3), 34.1 and 35.0 (C(CH3)3), 59.6 (NCH2), 117.7, 126.0, 127.1, 129.2, 129.6, 132.9, 136.7, 140.1, 158.0 (Ar–C) 166.9 (NCH). (77Se{1H} NMR, CDCl3): δ (ppm) = 281.1.
Synthesis of L2. The selenated amine C6H5Se–(CH2)2–NH2 (0.412 g, 2.06 mmol) and 2-(benzyloxy)-3,5-di-tert-butylbenzaldehyde (0.650 g, 2.00 mmol) were reacted in absolute ethanol (30 mL) at room temperature for 12 h. The volatiles were removed using rotary evaporator which resulted in yellow liquid. The yellow liquid formed was reacted with NaBH4 (0.080 g, 2.11 mmol) in ethanol to obtain ligand L2 as colourless viscous liquid in 91% yield (0.925 g, 1.82 mmol). 1H NMR (CDCl3): δ (ppm) = 1.32 (s, 9H, C(CH3)3), 1.43 (s, 9H, C(CH3)3), 1.74 (broad s, 1H, NH), 2.86 (t, JHH = 7.05 Hz, 2H, SeCH2), 2.97 (t, JHH = 6.45 Hz, 2H, NCH2CH2), 3.82 (s, 2H, CH2N), 5.00 (s, 2H, OCH2), 7.16–7.49 (broad m, 12H, Ar–H). 13C{1H} (CDCl3): δ (ppm) = 28.3 (CH2Se), 31.2 and 31.5 (C(CH3)3), 34.4 and 35.3 (C(CH3)3), 48.7 (NHCH2), 48.8 (CH2NH), 75.3 (OCH2), 123.2, 125.2, 126.4, 126.6, 127.3, 128.3, 128.8, 129.8, 132.5, 132.8, 138.0, 141.8, 145.8 and 154.1 (Ar–C). (77Se{1H} NMR, CDCl3): δ (ppm) = 267.8.
Synthesis of complex 1. The Na2PdCl4 (0.147 g, 0.5 mmol) was dissolved in 20 mL of methanol. The homogeneous methanolic solution of ligand L1 (0.208 g, 0.5 mmol dissolved in 10 mL of methanol) was added with stirring. The mixture was further stirred for 6 h. The orange red coloured precipitate was obtained which was filtered and dried under vacuo. The single crystals of 1 were grown from CH3OH–CH3CN mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) by slow evaporation of its solution for one week. Yield: 88% (0.245 g, 0.44 mmol). Anal. Calcd for C23H30ClNOPdSe (Mw: 557.29): C, 49.57; H, 5.43; N, 2.51%. Found: C, 49.60; H, 5.24; N, 2.64. 1H NMR (DMSO-d6): δ (ppm) = 1.23 (s, 9H, C(CH3)3), 1.37 (s, 9H, C(CH3)3), 3.10–3.20 (m, 2H, CH2), 3.72 (m, 1H, CH2), 4.46–4.50 (m, 1H, CH2), 7.17–8.18 (broad m, 5H, C6H5), 8.09 (s, 1H, NCH), 7.35 (s, 1H, C5H2), 8.19 (s, 1H, C5H2). 13C{1H} (CDCl3): δ (ppm) = 29.9 and 31.7 (C(CH3)3), 32.9 (CH2Se), 34.0 and 35.8 (C(CH3)3), 65.7 (NCH2), 119.3, 125.6, 128.7, 129.9, 130.4, 130.5, 133.5, 136.1, 138.8, 161.3 (Ar–C), 162.4 (NCH). (77Se{1H} NMR, CDCl3): δ (ppm) = 432.1.
:1) by slow evaporation of its solution for one week. Yield: 88% (0.245 g, 0.44 mmol). Anal. Calcd for C23H30ClNOPdSe (Mw: 557.29): C, 49.57; H, 5.43; N, 2.51%. Found: C, 49.60; H, 5.24; N, 2.64. 1H NMR (DMSO-d6): δ (ppm) = 1.23 (s, 9H, C(CH3)3), 1.37 (s, 9H, C(CH3)3), 3.10–3.20 (m, 2H, CH2), 3.72 (m, 1H, CH2), 4.46–4.50 (m, 1H, CH2), 7.17–8.18 (broad m, 5H, C6H5), 8.09 (s, 1H, NCH), 7.35 (s, 1H, C5H2), 8.19 (s, 1H, C5H2). 13C{1H} (CDCl3): δ (ppm) = 29.9 and 31.7 (C(CH3)3), 32.9 (CH2Se), 34.0 and 35.8 (C(CH3)3), 65.7 (NCH2), 119.3, 125.6, 128.7, 129.9, 130.4, 130.5, 133.5, 136.1, 138.8, 161.3 (Ar–C), 162.4 (NCH). (77Se{1H} NMR, CDCl3): δ (ppm) = 432.1.
Synthesis of 2. The Na2PdCl4 (0.147 g, 0.5 mmol) was dissolved in 5.0 mL of water. A solution of ligand L2 (0.254 g, 0.5 mmol) made in 10 mL of acetone was added to homogenized aqueous solution of Na2PdCl4 drop wise with vigorous stirring. The mixture was further stirred for 2 h resulting orange-red precipitate. It was poured into cold water (40 mL) and extracted with chloroform (4 × 30 mL). The combined extract was dried over anhydrous sodium sulphate. Its solvent was reduced to 5 mL and complex was precipitated using n-hexane. The precipitate was filtered and dried under vacuo. The single crystals of 2, as 2·CH3CN were grown from CH3OH–CH3CN mixture (1:1) by slow evaporation of its solution for one week. Yield: 84% (0.306 g, 0.42 mmol). Anal. Calcd for C30H38Cl2NOPdSe·CH3CN (Mw: 725.93): C, 52.94; H, 5.69; N, 3.86%. Found: C, 52.82; H, 5.67; N, 3.78. 1H NMR (CDCl3): δ (ppm) = 1.35 (s, 9H, C(CH3)3), 1.39 (s, 9H, C(CH3)3), 2.63–2.65 (m, 2H, CH2), 3.55–3.58 (broad m, 2H, CH2), 4.19–4.27 (m, 1H, CH2), 4.69–4.73 (m, 1H, CH2), 4.80–4.90 (m, 2H, OCH2), 7.20–7.87 (broad m, 12H, Ar–H). 13C{1H} (CDCl3): δ (ppm) = 29.3 and 29.7 (C(CH3)3), 29.6 and 33.4 (C(CH3)3), 33.0 (CH2Se), 33.5 (NHCH2), 49.0 (NH(CH2)Ar), 51.0 (OCH2Ph), 123.4, 124.5, 125.4, 125.6, 125.9, 126.6, 126.8, 128.1, 128.2, 131.2, 135.1, 140.0, 145.9, 152.9 (Ar–C). (77Se{1H} NMR, CDCl3): δ (ppm) = 489.3.
Procedure for Suzuki reaction of aryl bromides with phenylboronic acid. An oven dried flask was charged with aryl bromide (1.0 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2.0 mmol) and ethanol (4.0 mL). The flask was placed on an oil bath at 80 °C under aerobic condition and the reaction mixture stirred until maximum conversion of aryl bromide to coupled product occurred, as revealed with NMR spectroscopy. The mixture was extracted with diethyl ether (100 mL). The extract was washed with water (100 mL) and dried over anhydrous Na2SO4.
Results and discussion
Synthesis
The synthetic details of ligands L1 and L2, and their corresponding palladium metal complexes 1 and 2 are summarized in Scheme 1. The L1, L2, 1 and 2·CH3CN are stable under ambient conditions. The complexes can be stored for six month without noticeable decomposition. The ligands and their complexes 1 and 2 have been characterized by their 1H and 13C{1H} and 77Se{1H} NMR spectra (see ESI† for the spectra). These data are consistent with the structures depicted for them in Scheme 1.
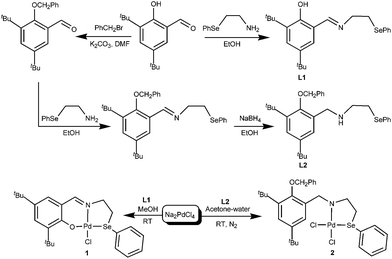 |
| | Scheme 1 Synthesis of ligands L1 and L2 and their Pd(II) complexes. | |
Crystal structures
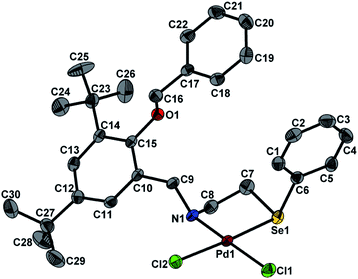
Molecular structures of 1 and 2 were determined with single crystal X-ray diffraction. The crystal and structure refinement data are given in Table 1. The Fig. 1 depicts molecular structure of 1 (ellipsoid at 30% probability level; hydrogen atoms have been omitted for clarity). The palladium in 1 is surrounded by O1, N1, Se1 and Cl1 resulting in almost square planner geometry as shown in Fig. 1. The ligand in complex 1, coordinates with Pd in a mono anionic tridentate (Se, N, O−) mode forming a six and five membered chelate ring. The six membered ring is formed via O− and N whereas five membered via Se and N. The Pd–N bond length 1.996(4) Å in 1 is comparable to the reported values of 1.985(4) Å for [PdCl{2–O–3–CH(CH2CH3)2–C6H3–CN–(CH2)2SeMe}] (I) and 2.003(7) Å for [PdCl{2–O–C6H4C(CH3)N(CH2)2SePh}] (II).4a,b However, it is slightly shorter than those reported for [PdCl{2–O––C6H4C(C6H5)N(CH2)2SePh}] (III) 2.010(4) Å and [PdCl{2–O–C10H6C(CH3)N(CH2)2SePh}] (IV) 2.010(3) Å.4c,d The Pd–O distance 2.001(3) Å in 1 is comparable with values reported for (I) 2.017(4) and (III) 1.993(3) Å and longer than those of (II) 1.977(6) and (IV) 1.973(2) Å. Similarly Pd–Se bond distance 2.370(1) Å in 1 is comparable with values for (I) 2.365(1) and (II) 2.367(1) Å and longer than those of (III) 2.358(1) and (IV) 2.360(1) Å. The Pd–Cl distance 2.302(1) Å in 1 is comparable with the value reported for (II) 2.305(2) Å and little shorter than those of (I) 2.323(2), (III) 2.315(2) and (IV) 2.316(1) Å. The molecular structure of 2 is shown in Fig. 2 (Ellipsoid at 30% probability level; hydrogen atoms have been omitted for clarity). The palladium in 2 has nearly square planner geometry constituted by N1, Se1, Cl1 and Cl2. The bond distances: Pd–Se 2.366(1) and Pd–Cl1 2.306(1) Å in 2 are comparable with the values mentioned above, 2.370(1) Å and 2.302(1) Å in case of 1, respectively. However, the Pd–N distance 1.996(4) Å in 1 is shorter than 2.066(3) Å found in case of 2·CH3CN. Significant non-covalent interactions observed in 1 and 2 are listed in Table 2. In the crystal of complex 1 intermolecular C–H⋯Cl non-covalent interactions exist as shown in Fig. 3. The Cl1 atom acts as a bifurcated hydrogen bond acceptor with H2 (SePh) and H8A (methylene group of adjacent molecule; C8). In crystal of 2·CH3CN C–H⋯N hydrogen bonding interactions are present in addition to intermolecular C–H⋯Cl non-covalent interactions as shown in Fig. 4. In this crystal, the Cl2 atom acts as a bifurcated hydrogen bond acceptor with H8B (methylene group; C8) and H20 (CH2Ph) of another adjacent molecule. The Cl1 atom is hydrogen bonded with H31B (CH3CN) of adjacent molecule. In addition, the N2 atom of CH3CN acts as hydrogen bond acceptor with H16A (C16 of CH2Ph group) of adjacent molecule.
 |
| | Fig. 1 ORTEP representation of 1. Selected bond lengths (Å) and angles (°): Pd1–N1 1.996(4), Pd1–O1 2.001(3), Pd1–Se1 2.370(1), Pd1–Cl1 2.302(1); N1–Pd1–O1 92.5(2), N1–Pd1–Cl1 178.3(1), O1–Pd1–Cl1 88.3(1), N1–Pd1–Se1 89.1(1), O1–Pd1–Se1 177.2(1), Cl1–Pd1–Se1 90.1(1). | |
 |
| | Fig. 2 ORTEP representation of 2. Selected bond lengths (Å) and angles (°): Pd1–N1 2.066(3), Pd1–Se1 2.366(1), Pd1–Cl1 2.306(1), Pd1–Cl2 2.331(1); N1–Pd1–Cl1 176.0(1), N1–Pd1–Cl2 88.0(1), Cl1–Pd1–Cl2 95.1(1), N1–Pd1–Se1 88.9(2), Cl1–Pd1–Se1 87.9(4), Cl2–Pd1–Se1 176.8(1). | |
Table 2 Selected non-covalent interactions of 1 and 2 (inter atomic distances in Å and bond angles in deg)
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
D–H⋯A |
| ½ − x, −1/2 + y, z; 1 − x, 1 − y, −z; 1 − x, 1 − y, 1 − z; x, y, 1 + z; − 1 + x, y, z; 1 − x, −y, 2 − z. |
| 1 |
| C8–H8A⋯Cl1a |
0.97 |
2.68 |
3.45 |
140 |
| C2–H2⋯Cl1b |
0.93 |
2.85 |
3.69 |
151 |
| |
| 2 |
| C16–H16A⋯N2c |
0.97 |
2.69 |
3.55 |
148 |
| C31–H31B⋯Cl1d |
0.96 |
2.79 |
3.71 |
162 |
| C20–H20⋯Cl2e |
0.93 |
2.90 |
3.61 |
135 |
| C8–H8B⋯Cl2f |
0.97 |
2.90 |
3.68 |
139 |
 |
| | Fig. 3 Packing diagram of 1 illustrating intermolecular C–H⋯Cl hydrogen bonding in the crystal lattice. | |
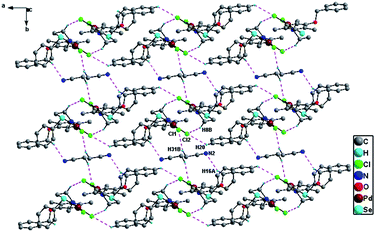 |
| | Fig. 4 Packing diagram of 2·CH3CN illustrating intermolecular C–H⋯Cl and C–H⋯N hydrogen bonding in the crystal lattice. | |
Spectroscopic studies
The 1H and 13C{1H} NMR spectra of ligand L1 and L2 have been found consistent with their structures depicted in Scheme 1. The signal of OH proton in 1H NMR spectrum of L1 has been observed at δ 13.55 ppm. The 77Se{1H} NMR spectrum of L1 has a signal at δ 281.1 ppm consistent with the value reported in literature (δ 281.5 ppm for selenoether4d). In 1H NMR spectrum of ligand L2, the signal of NH proton appears as a broad singlet at δ 1.74 ppm. The signals for CH2N and OCH2 protons in proton NMR spectrum of ligand L2 appear at δ 3.82 and 5.00 ppm, respectively. All expected signals were observed in 13C{1H} NMR spectra of ligands L1 and L2. 77Se{1H} NMR spectrum L2 shows a signal at δ 268.9 ppm. In complex 1, the ligand L1 coordinates to Pd in a mono anionic tridentate (Se, N, O−) mode which is corroborated by its single crystal structure. The signal of phenolic proton present at δ = 13.55 ppm in the NMR spectrum of ligand L1 has been found disappeared on complexation. This indicates the deprotonation of phenolic OH on formation of complex 1. In 1H NMR spectrum of complex 1, each proton of both CH2 groups becomes diastereotopic and this results in three multiplets at δ, 3.10–3.20, 3.72 and 4.46–4.50 ppm (2H, 1H and 1H, respectively; see Fig. S7 in ESI†), supporting Pd–N and Pd–Se bond formation. The 77Se{1H} NMR spectrum of complex 1 has a signal at δ 432.1 ppm which is highly deshielded (151 ppm) compared to that of free ligand. This may be ascribed due to coordination of selenium to palladium. The large deshielding may be due to formation of five membered chelate ring4d with Pd. In complex 2 the ligand L2 coordinates to Pd in a neutral bidentate (Se, N) mode as corroborated by its single crystal structure. In 1H NMR spectrum of complex 2 also, protons of each CH2 group become diastereotopic and result in five multiplets at δ 2.63–2.65, 3.55–3.58, 4.19–4.27, 4.69–4.73 and 4.80–4.90 ppm (2H, 2H, 1H, 1H, and 2H, respectively see Fig. S10 in ESI†), consistent with Pd–N and Pd–Se bond formation, which makes the NCH2 and CH2Se protons rigid. The 77Se{1H} NMR spectrum of complex 2 has a signal at δ 489.3 ppm, highly deshielded (221.5 ppm) compared to that of free ligand, indicating the coordination of selenium with palladium.
Suzuki–Miyaura C–C coupling catalyzed with 1 and 2
Suzuki–Miyaura C–C coupling reactions were carried out in the presence of complexes 1 and 2 as catalyst and results obtained are summarized in Tables 3 and 4. The optimization of reaction conditions with present catalyst 1 was carried out by coupling 4-bromotoulene and phenylboronic acid under aerobic conditions at 80 °C for 5 h using different bases and solvents. The best results were obtained with K2CO3 and ethanol (Table 3, entry 6). The conversion was also observed in water (up to 40%) with K2CO3 (Table 3, entry 13). The coupling reaction of 4-bromo-nitrobenzene/4-bromobenzo-nitrile/4-bromoacetophenone with phenylboronic acid in the presence of 0.1 mol% of 1 or 2 in 5 h at 80 °C, resulted in corresponding biaryl in 100% yield (Table 4, entry 1–6). The coupling between 4-bromotoluene and phenylboronic acid in the presence of 0.1 mol% of 1 for 5 h at 80 °C, resulted in corresponding biaryl in 88% yield (Table 4, entry 7). However, when catalyst 2 was used for the same coupling under similar reaction conditions yield of corresponding coupling product, biaryl reduced to 82% (Table 4, entry 8). Similar trends were observed in the case of 4-bromobenzaldehyde, 4-bromoanisole and 4-bromobenzoic acid under similar reaction condition which gave 75, 35 and 70% conversion to corresponding biaryl with catalyst 1 and 71, 32 and 61% with catalyst 2. These trends are due to the fact that the catalytic activity is dependent on the nature of the electron withdrawing group on the aryl ring, the reactivity increases in the order of NO2 > H > OMe. The catalytic efficiency of 1 appears to be slightly higher than that of 2. In comparison to palladium complexes4a,c–e,9,11 other than the palladacycles and pincer ligand based ones the performance of 1 and 2 is comparable or better in comparison to those containing N/S donors. Similarly the present complexes are comparable (in some cases favourably) with palladium nanoparticle based catalytic systems.28 Air and moisture insensitivity of 1 and 2 are their additional advantages. However they cannot be recycled for catalytic applications.
Table 3 Optimization of reaction conditions for Suzuki–Miyaura C–C coupling reactions of 4-bromotoluene with phenylboronic acida
| Entry no |
Solvent |
Base |
Yieldb (%) |
| Reaction conditions: 1.0 equiv. of aryl halide, 1.2 equiv. of phenylboronic acid, and 2 equiv. of base and temperature of bath 80 °C. Catalyst 1: 0.1 mol% Pd. Time: 5 h. NMR(%) yield. |
| 1 |
1,4-Dioxane (4 mL) |
CH3ONa |
42 |
| 2 |
1,4-Dioxane (4 mL) |
K2CO3 |
50 |
| 3 |
DMF (4 mL) |
CH3ONa |
41 |
| 4 |
DMF (4 mL) |
K2CO3 |
47 |
| 5 |
EtOH (4 mL) |
CH3ONa |
82 |
| 6 |
EtOH (4 mL) |
K2CO3 |
88 |
| 7 |
1,4-Dioxane:water (3:1 mL) |
CH3ONa |
41 |
| 8 |
1,4-Dioxane:water (3:1 mL) |
K2CO3 |
42 |
| 9 |
DMF:water (3:1 mL) |
CH3ONa |
33 |
| 10 |
DMF:water (3:1 mL) |
K2CO3 |
39 |
| 11 |
EtOH:water (3:1 mL) |
CH3ONa |
52 |
| 12 |
EtOH:water (3:1 mL) |
K2CO3 |
68 |
| 13 |
Water (4 mL) |
K2CO3 |
40 |
Table 4 Suzuki–Miyaura coupling reaction catalyzed by catalysts 1 and 2a

|
| Entry no |
Aryl halide |
Catalyst |
Yieldb (%) (TON) |
TOF (h−1) |
| Reaction conditions: 1.0 equiv. of aryl halide, 1.2 equiv. of phenylboronic acid, and 2 equiv. of base (K2CO3), solvent: EtOH (4 mL) and temperature of bath 80 °C. Catalyst: 0.1 mol% Pd; time: 5 h. NMR(%) yield. |
| 1 |
4-Bromonitrobenzene |
1 |
100(1000) |
200 |
| 2 |
4-Bromonitrobenzene |
2 |
100(1000) |
200 |
| 3 |
4-Bromobenzonitrile |
1 |
100(1000) |
200 |
| 4 |
4-Bromobenzonitrile |
2 |
100(1000) |
200 |
| 5 |
4-Bromoacetophenone |
1 |
100(1000) |
200 |
| 6 |
4-Bromoacetophenone |
2 |
100(1000) |
200 |
| 7 |
4-Bromotoluene |
1 |
88(880) |
176 |
| 8 |
4-Bromotoluene |
2 |
82(820) |
164 |
| 9 |
4-Bromobenzaldehyde |
1 |
75(750) |
150 |
| 10 |
4-Bromobenzaldehyde |
2 |
71(710) |
142 |
| 11 |
4-Bromoanisol |
1 |
35(350) |
70 |
| 12 |
4-Bromoanisol |
2 |
32(320) |
64 |
| 13 |
4-Bromobenzoic acid |
1 |
70(700) |
140 |
| 14 |
4-Bromobenzoic acid |
2 |
61(610) |
122 |
Conclusions
Two sterically hindered selenated Schiff bases and their palladium(II) complexes have been synthesized and structurally characterized with 1H, 13C{1H} and 77Se{1H} NMR spectra. In 77Se{1H} NMR spectra deshielding of signal on complexation was up to 221.5 ppm compared to that of corresponding free ligand. The ligand L1 coordinates as tridentate (N, Se, O−) mode in complex 1 whereas L2 coordinates as a bidentate (N, Se) ligand in 2 as revealed by single crystal structure. Complexes 1 and 2 are efficient catalysts for Suzuki–Miyaura C–C coupling reactions for aryl bromide with phenylboronic acid in presence of K2CO3 in ethanol at 80 °C in 5 h. The reactivity increases on the nature of the substituent on the aryl ring and follows the order NO2 > H > OMe. The catalytic conversion was also observed up to 40% with K2CO3 in water with catalyst 1. The efficiency of 1 in carrying out the coupling is slightly higher than that of 2.
Acknowledgements
The authors acknowledge the Department of Science and Technology, New Delhi (SR/FT/CS-79/2011) (UK) and (SR/WOS-A/CS-57/2012) (VVS) for financial support.
Notes and references
- A. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 11949–11977 RSC.
-
(a) W.-G. Jia, Y.-B. Huang, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2008, 5612–5620 RSC;
(b) W.-G. Jia, Y.-B. Huang, Y.-J. Lin, G.-L. Wang and G.-X. Jin, Eur. J. Inorg. Chem., 2008, 4063–4073 CrossRef CAS PubMed.
-
(a) Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997–2999 CrossRef CAS PubMed;
(b) J. Aydin, N. Selander and K. J. Szabó, Tetrahedron Lett., 2006, 47, 8999–9001 CrossRef CAS PubMed;
(c) D. Das, G. K. Rao and A. K. Singh, Organometallics, 2009, 28, 6054–6058 CrossRef CAS;
(d) D. Das, P. Singh, M. Singh and A. K. Singh, Dalton Trans., 2010, 10876–10882 RSC.
-
(a) I. D. Kostas, B. R. Steele, A. Terzis, S. V. Amosova, A. V. Martynov and N. A. Makhaeva, Eur. J. Inorg. Chem., 2006, 2642–2646 CrossRef CAS PubMed;
(b) A. Kumar, M. Agarwal and A. K. Singh, Polyhedron, 2008, 27, 485–492 CrossRef CAS PubMed;
(c) A. Kumar, M. Agarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533–3545 CrossRef CAS PubMed;
(d) A. Kumar, M. Agarwal and A. K. Singh, Inorg. Chim. Acta, 2009, 362, 3208–3218 CrossRef CAS PubMed;
(e) G. K. Rao, A. Kumar, B. Kumar, D. Kumar and A. K. Singh, Dalton Trans., 2012, 1931–1937 RSC;
(f) P. Singh and A. K. Singh, Organometallics, 2010, 29, 6433–6442 CrossRef CAS;
(g) P. Singh and A. K. Singh, Inorg. Chim. Acta, 2012, 387, 441–445 CrossRef CAS PubMed.
- M. Mešková and M. Putala, Tetrahedron: Asymmetry, 2013, 24, 894–902 CrossRef PubMed.
- W. Tang, A. G. Capacci, X. Wei, W. Li, A. White, N. D. Patel, J. Savoie, J. J. Gao, S. Rodriguez, B. Qu, N. Haddad, B. Z. Lu, D. Krishnamurthy, N. K. Yee and C. H. Senanayake, Angew. Chem., Int. Ed., 2010, 49, 5879–5883 CrossRef CAS PubMed.
-
(a) M. Austeri, M. Enders, M. Nieger and S. Bräse, Eur. J. Org. Chem., 2013, 1667–1670 CrossRef CAS PubMed;
(b) L. Monnereau, D. Sémeril, D. Matt and L. Toupet, Chem.–Eur. J., 2010, 16, 9237–9247 CrossRef CAS PubMed;
(c) J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746–4748 CrossRef CAS PubMed.
-
(a) M. G. Organ, S. Çalimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem., Int. Ed., 2009, 48, 2383–2387 CrossRef CAS PubMed;
(b) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed;
(c) C. J. O'Brien, E. A. B. Kantchev, C. Valente, N. Hadei, G. A. Chass, A. Lough, A. C. Hopkinson and M. G. Organ, Chem.–Eur. J., 2006, 12, 4743–4748 CrossRef PubMed;
(d) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed;
(e) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS PubMed;
(f) C. W. K. Gstöttmayr, V. P. W. Böhm, E. Herdtweck, M. Grosche and W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1363–1365 CrossRef.
- A. Bermejo, A. Ros, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2008, 130, 15798–15799 CrossRef CAS PubMed.
-
(a) G. K. Rao, A. Kumar, S. Kumar, U. B. Dupare and A. K. Singh, Organometallics, 2013, 32, 2452–2458 CrossRef CAS;
(b) G. K. Rao, A. Kumar, J. Ahmedz and A. K. Singh, Chem. Commun., 2010, 46, 5954–5956 RSC;
(c) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, E. I. Gutsul, A. A. Vasil'ev, P. V. Petrovskii and I. L. Odinets, Dalton Trans., 2009, 8657–8666 RSC;
(d) S. M. Nobre and A. L. Monteiro, J. Mol. Catal. A: Chem., 2009, 313, 65–73 CrossRef CAS PubMed;
(e) D. Zim, S. M. Nobre and A. L. Monteiro, J. Mol. Catal. A: Chem., 2008, 287, 16–23 CrossRef CAS PubMed;
(f) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, E. I. Gutsul, L. N. Puntus, A. A. Vasil'ev, P. V. Petrovskii and I. L. Odinets, Organometallics, 2008, 27, 4062–4070 CrossRef CAS;
(g) Z. Xiong, N. Wang, M. Dai, A. Li, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3337–3340 CrossRef CAS PubMed.
-
(a) K. N. Sharma, H. Joshi, V. V. Singh, P. Singh and A. K. Singh, Dalton Trans., 2013, 3908–3918 RSC;
(b) F. Saleem, G. K. Rao, A. Kumar, P. Singh and A. K. Singh, Organometallics, 2013, 32, 387–395 CrossRef CAS;
(c) C. Fliedel and P. Braunstein, Organometallics, 2010, 29, 5614–5626 CrossRef CAS;
(d) D. Yuan and H. V. Huynh, Organometallics, 2010, 29, 6020–6027 CrossRef CAS;
(e) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, A. A. Vasil'ev, P. V. Petrovskii and I. L. Odinets, Organometallics, 2010, 29, 2054–2062 CrossRef CAS.
-
(a) K. N. Sharma, H. Joshi, A. K. Sharma, O. Prakash and A. K. Singh, Organometallics, 2013, 32, 2443–2451 CrossRef CAS.
-
(a) Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed;
(b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. de Meijere, Wiley-Interscience, New York, 2002, p. 1669 Search PubMed;
(c) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998 Search PubMed.
-
(a) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed;
(b) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed;
(c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed;
(d) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS PubMed.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
-
(a) F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed;
(b) S. Santra, P. K. Hota, R. Bhattacharyya, P. Bera, P. Ghosh and S. K. Mandal, ACS Catal., 2013, 3, 2776–2789 CrossRef CAS;
(c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed;
(d) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed;
(e) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Khanna, A. Bala and B. L. Khandelwal, J. Organomet. Chem., 1995, 494, 199–204 CrossRef CAS.
- C. Costentin, M. Robert, J.-M. Savéant and C. Tard, Angew. Chem., Int. Ed., 2010, 49, 3803–3806 CrossRef CAS PubMed.
- SMART, Bruker Molecular Analysis Research Tool, version 5.0, Bruker Analytical X-ray Systems, Madison, WI, 2000 Search PubMed.
- ENHANCE, Oxford Xcalibur Single Crystal Diffractometer, version 1.171.34.40, Oxford Diffraction Ltd, Oxford, U.K., 2006 Search PubMed.
- CrysAlisPro, version 1.171.34.40, Oxford Diffraction Ltd, Oxford, U.K., 2006 Search PubMed.
- SAINT-NT, version 6.04, Bruker Analytical X-ray Systems, Madison, WI, 2001 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- K. Brandenburg, DIAMOND, version 2.0 c, University of Bonn, Bonn, Germany, 2004 Search PubMed.
-
(a) V. V. Singh, U. Kumar, S. N. Tripathi and A. K. Singh, Dalton Trans., 2014, 12555–12563 RSC;
(b) M. Zhu, Y. Wang, C. Wang, W. Lia and G. Diao, Catal. Sci. Technol., 2013, 3, 952–961 RSC;
(c) M. Zhu and G. Diao, J. Phys. Chem. C, 2011, 115, 24743–24749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 77Se{1H} NMR spectra of L1, L2, 1 and 2. CCDC 1009785 and 1009786. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra06313a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.