
The required level of isosteric heat for the adsorptive/storage delivery of H2 in the UiO series of MOFs†
K. Vasanth Kumar*,
G. Charalambopoulou,
M. Kainourgiakis,
A. Gotzias,
A. Stubos and
Th. Steriotis
National Center for Scientific Research “Demokritos”, Agia Paraskevi Attikis, 15310, Athens, Greece. E-mail: vasanth_vit@yahoo.com
First published on 27th August 2014
Abstract
The required level of isosteric heat of adsorption for efficient storage and delivery of H2 in the UiO series of MOFs was theoretically predicted using molecular simulations. Very high isosteric heats may lead to enhanced storage capacities, however the real H2 delivery capacity is practically reduced. In this respect, for maximum H2 delivery, there exists an optimum isosteric heat value (28–29 kJ mol−1).
Storage of hydrogen is a bottleneck in shifting our society from the use of fossil fuels to the hydrogen economy. In recent years adsorption in porous materials has been considered to be safe and efficient for this purpose as the conventional storage methods such as cryogenic storage of hydrogen or high-pressure compression operate at extreme and impractical conditions.1,2 Amongst others, metal–organic frameworks (MOFs) are considered to be promising adsorbents for H2 storage due to their available surface areas and porosity.3,4 The newly emerging UiO type MOFs are receiving great attention due to their remarkable stability to temperature, pressure and humid conditions.5–8
The ‘ultimate full fleet target’ of United States Department of Energy (DOE) requires a H2 gravimetric storage capacity of 7.5 wt% and a volumetric storage capacity of 70 g L−1 on a system basis at the earliest possible date (the respective short-term targets have been set to 4.5 wt% and 36 g H2 per L for 2010 and 5.5 wt% and 40 g L−1 for 2017).9 If the UiO series of MOFs could meet some of these targets at room temperature, then their enhanced stability would give hope and motivation to implement them in real on-board H2 storage tanks. At room temperature the adsorption forces between H2 and the MOF atoms are nearly equal to the thermal motions of the H2 gas molecules; thus the respective adsorption capacity is usually very low and far from the above targets. To overcome this limitation and improve the room temperature adsorption capacity, experimentalists focus on functionalizing the linker molecules with different groups or decorating the framework with metals and/or heteroatoms such as titanium, lithium, iron, vanadium, boron, nitrogen etc.10–13 Even minute quantities of such dopants may have a profound effect on the electron density of the linker molecules and consequently may increase the framework-H2 interactions.
In this respect, a tremendous amount of work has been invested in recent years, in obtaining UiO type MOFs with different surface chemistry, that mainly targets to improve their storage capacity for H2 (but also other gases), as well as the respective isosteric heat of adsorption, Qst.14–16 In principle Qst is a function of coverage however in this framework the interest is focused at “zero coverage”, i.e. the heat of adsorption at infinite dilution, which is directly related to the H2-framework interactions. Previously, Bae and Snurr17 demonstrated that improving Qst of high surface (>4800 m2 g−1) area porous materials is one possible strategy to achieve the DOE targets. Theoretically, by increasing Qst, the H2 capacity increases at lower pressures. However, for practical applications, the amount stored at the discharge pressure is equally important to maximum H2 storage capacity as the ultimate delivery performance depends on both charging and discharging capacities (nevertheless deliverable capacity is rarely used as a criterion for assessing the H2 storage performance of MOFs). Thus the goal is to find the optimal Qst that can give high adsorption capacities without penalizing the deliverable capacity, defined as the amount of hydrogen adsorbed at high-pressure (e.g. 120 bar) minus the amount adsorbed at the discharge pressure (near-ambient). As far as UiO MOFs are concerned, until now the required level of isosteric heat for the storage or delivery of H2 is still not clearly understood. Thus in this work we examine the level of H2/MOF interactions needed for efficient storage and delivery of H2 as well as the conditions required to meet the DOE targets with this particular series of MOFs.
To do this, we used molecular simulations to estimate the optimum level of isosteric heat of adsorption for the room temperature storage of H2 up to 120 bar and delivery at 1 bar (in consistency with the DOE target requirements) in three UiO type MOFs, UiO-66, UiO-67 and UiO-68.
Grand Canonical Monte Carlo (see ESI†) simulations were performed to obtain the H2 adsorption isotherms. As already mentioned, the usual experimental strategies for improving the storage capacity of MOFs at room temperature involve functionalization or doping of linker molecules. The net effect is expected to be an enhanced solid–fluid interaction (and thus isosteric heat) and ultimately higher adsorption at room temperature than that observed with the parent MOF structures. Theoretically, the simulation of such a scenario could be carried out in terms of an average increased solid–fluid interaction in a rigid framework. This is the essence of the so-called “chemisorption” model discussed by Bae and Snurr17 for MOFs as well as Cracknell18 and Wang and Johnson,19 for other classes of porous materials. In this work we modified the isosteric heat by artificially increasing the Lennard-Jones (LJ) parameter epsilon between the atoms on the linker molecules, La (carbon and hydrogen atoms), of the UiO MOFs and H2. More specifically, the potential well depth, i.e., the epsilon parameter between H2/linker-atoms, εLa/H2, was arbitrarily multiplied by 2, 3, 4, 5, 6, and 7; hereafter referred to as chemisorption potentials. Such an assumption can be considered as an analogue to the increase in the overall binding energy of the material via a doping or functionalization process. Such processes can significantly alter the pore topology, surface area and pore volume of a certain MOF structure. In addition, the presence of even minute amount of heavy metals or other dopants will also change the framework density thus altering the ultimate gravimetric storage capacity. By artificially scaling H2/framework interactions it is possible to systematically study the sole effect of Qst on the H2 storage and deliverable capacity of UiO type MOFs without altering their pore properties. Complete details about the simulation methodology and the potential parameters used and the strategies adopted are given in the ESI.† The adsorption isotherms are discussed in terms of adsorption excess, a quantity that can be measured through experiments.
Before studying the adsorption in the UiO series of MOFs, we characterized their pore properties in terms of surface area and accessible pore volume by using a geometrical method (see ESI†). The crystallographic density, calculated surface area and pore volume values are given in Table 1. The calculated pore volume and specific surface area for UiO-66 and UiO-67 are in good agreement with experimental values reported in literature.5 In addition, we also estimated the pore size distribution of these MOFs using pore-blazer v3.0 which is based on the geometric method, originally proposed and developed by Sarkisov and Harrison.20 The pore size distribution results show that UiO-66 essentially contains ultra-micropores that range from 6.625 to 8.125 Å. UiO-67 and UiO-68 contain larger micropores that range from 9.125–12.375 Å and 12.375–16.875 Å, respectively. The pore size distribution plot is given in the ESI.†
| MOF | Surface area, m2 g−1 | Vtotal, cm3 g−1 | ρc, g cm−3 |
|---|---|---|---|
| UiO-66 | 840 | 0.475 | 1.215 |
| UiO-67 | 3008 | 0.99 | 0.708 |
| UiO-68 | 4162 | 1.707 | 0.462 |
Storage of H2 in UiO MOFs
In Fig 1 we present the H2 gravimetric and volumetric storage capacity of UiO-66, UiO-67 and UiO-68 at 298 K as a function of pressure, as obtained from simulations after using both the actual and scaled epsilon parameters. It should be noted that the volumetric storage values were obtained by assuming that the packing density is equivalent to the crystallographic density and thus the reported values herein correspond to the upper theoretical limit of the studied MOFs. It is clear from Fig. 1 that the H2 storage capacity of the studied MOFs at room temperature are extremely low throughout the range of pressures studied if the adsorption is only due to dispersion forces. For instance, UiO-66 could store only up to 0.18 wt% at 100 bar by physisorption; UiO-67 and UiO-68 can store 0.33 wt% and 0.43 wt% at 100 bar, respectively. This is an expected result as H2 is supercritical at 298 K and adsorption is governed by the high thermal motions of H2 and the weak fluid–fluid interactions.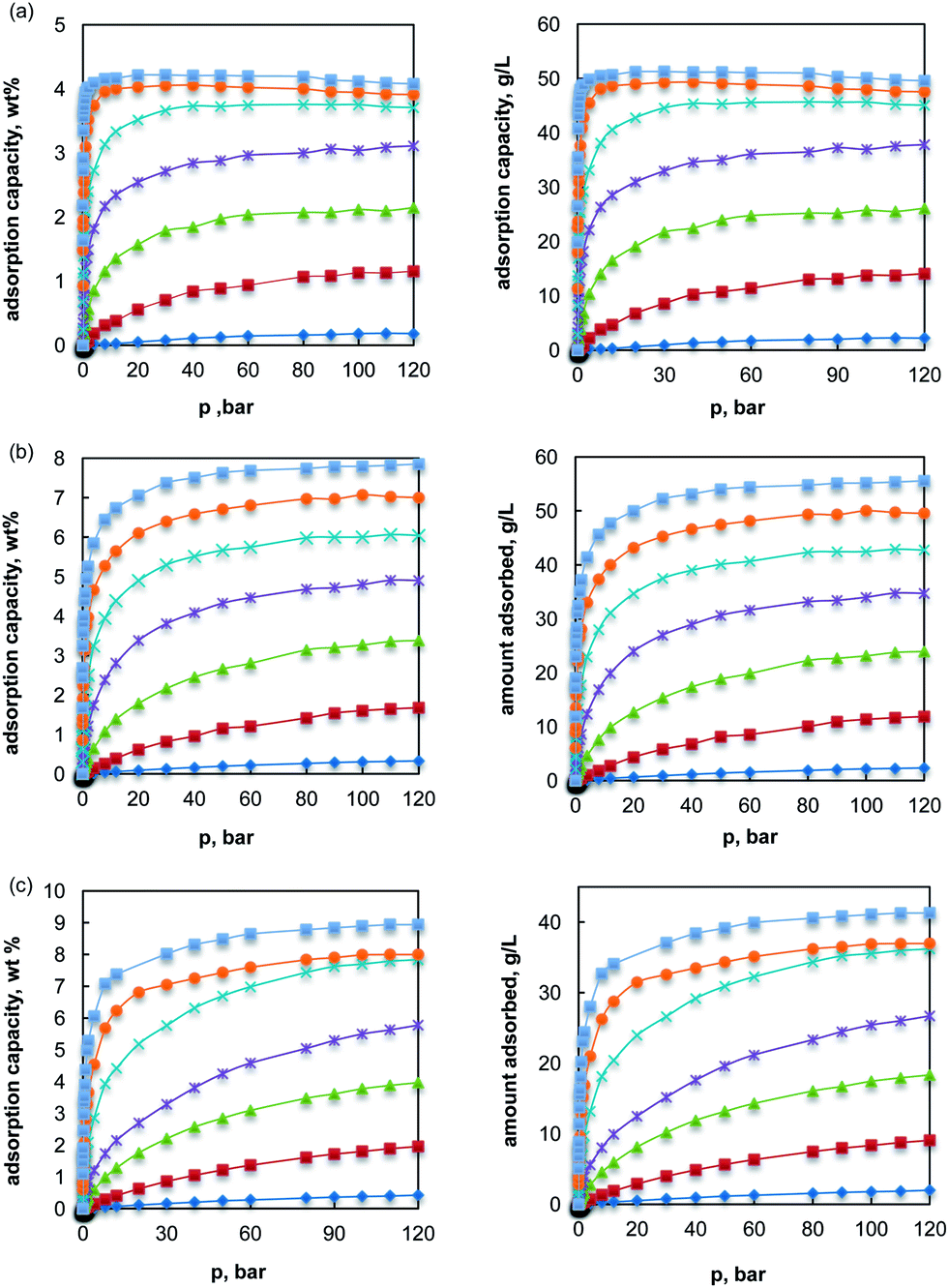 | ||
Fig. 1 Adsorption isotherms for hydrogen molecules in UiO series of MOFs with physisorption and chemisorption potentials at 298 K (a) UiO-66, (b) UiO-67 and (c) UiO-68 (left panel: gravimetric storage capacity; right panel: volumetric storage capacity) ( : εLa/H2; : εLa/H2; : 2εLa/H2; : 2εLa/H2; : 3εLa/H2; : 3εLa/H2; : 4εLa/H2; : 4εLa/H2; : 5εLa/H2; : 5εLa/H2; : 6εLa/H2; : 6εLa/H2; : 7εLa/H2). : 7εLa/H2). | ||
In all the three studied structures, the increase of the isosteric heat by scaling the εLa/H2 interactions revealed a pronounced effect on the H2 storage capacity throughout the range of pressures studied. For instance, even a two-fold increase in the solid–fluid potential between the linker molecule and H2, has drastically increased the maximum excess adsorption capacity of UiO-66 from 0.18 wt% to 1.15 wt% (or 14 g L−1), while by further increasing the εLa/H2 potential seven times (7εLa/H2) the storage capacity increased to 4.07 wt% (or 49 g L−1). It should be noted that in UiO-66, that contains ultra-micropores, the enhancement of dispersion forces by five or more times also reduces the pressure at which the maximum excess adsorption occurs. For example, by increasing the εLa/H2 potential by a factor of five (or more) the pressure at which the excess adsorption reaches a maximum was significantly reduced (40 bar). Although the volumetric storage value of 49 g L−1 surpasses the 2010 target, the respective gravimetric storage capacity of this structure is slightly lower than the 2010 target and far away from the 2017 or the ultimate full fleet target. Thus, improving the Qst of the UiO-66 which has low pore volume and surface area might not be the correct strategy towards achieving simultaneously the gravimetric and volumetric storage capacity targets set by DOE.
In the case of UiO-67, increasing the solid–fluid potential energy from 2εLa/H2 to 7εLa/H2 causes a significant enhancement in the gravimetric storage capacity from 1.67 wt% (or 11.85 g L−1) to 7.85 wt% (or 55.57 g L−1) at 298 K and 100 bar. In UiO-68 that contains pores of relatively larger size, increasing the potential from 2εLa/H2 to 7εLa/H2 increased the storage capacity from 1.95 wt% (or 9.04 g L−1) to 8.94 wt% (or 41.28 g L−1).
According to the results presented in Fig. 1, it can be seen that if the εLa/H2 interactions become seven times larger compared to the actual values, the gravimetric storage capacity for UiO-67 and UiO-68, that contains larger pore volume and surface area, can meet the ultimate full fleet gravimetric storage target as well as both the 2017 gravimetric and volumetric storage capacity targets. This is a useful observation as none of the previous experimental or theoretical studies on MOFs have proposed a feasible scenario that could lead simultaneously to both high volumetric and gravimetric storage capacity. These results are also supported by the snapshots of Fig. 2 that illustrates the H2 adsorption in UiO-67 structure by physisorption and a chemisorption potential (7εLa/H2). Our findings indicate that in order to achieve the desired gravimetric and volumetric storage capacity a mere increase of the isosteric heat is not enough as the porous materials under consideration should have a certain combination of surface area and pore volume. For instance when strong chemisorption is assumed (εLa/H2 = 7εLa/H2), the volumetric storage capacity of UiO-68 is predicted to be considerably lower than that of UiO-67, although the surface area of the first structure is significantly higher than the second one. Thus based on the present results, the surface area (∼3000 m2 g−1) and pore volume (∼1 cm3 g−1) of UiO-67 can be considered as an approximate reference when selecting the starting porous material to be used in a design strategy aiming to the improvement of the isosteric heat of adsorption. However, such a material might not be the optimum basis for practical applications if the actual H2 deliverable capacity is also considered.
 | ||
| Fig. 2 Snapshots showing H2 adsorption in UiO-67 MOF: (a) physisorption and (b) chemisorption with εa/H2 = 7εLa/H2. | ||
Delivery of H2 with UiO MOFs
The design of hydrogen storage materials requires that the system both charges and discharges rapidly and completely under near-ambient conditions. Thus, any attempt to improve Qst should be carefully designed in a way that the respective material retains only a small amount of H2 at the delivery pressure. To find how the isosteric heat affects the H2 deliverable capacity of the UiO series of MOFs, we plotted in Fig. 3 the difference between the amounts adsorbed at 120 bar and 1 bar (as calculated with all 6 chemisorption potentials assumed) as a function of the predicted average isosteric heat of adsorption (calculated at pressures up to 1 bar). Fig. 3 clearly shows that there exists an optimum isosteric heat value at which the deliverable capacity is maximum or in other words there exists a point of compensation at optimal Qst between the maximum adsorption at the high charging pressure and the minimum adsorption at the low discharging pressure. Interestingly this optimal isosteric heat seems to depend on the porous nature of the studied MOFs. For UiO-66 that contains ultra-micropores, this optimal isosteric heat is around 21 kJ mol−1 while in the case of UiO-67 and UiO-68 that contain larger micropores, the optimal isosteric heat is slightly higher i.e. around 28–29 kJ mol−1. This is in close agreement with the earlier work of e.g. Bae and Snurr17 who reported that regardless the MOF pore structure or surface chemistry, the optimal value of Qst falls in the range 23–28 kJ mol−1. In addition, it is generally assumed that during physisorption of H2, pore volume and surface area are correlated to volumetric and gravimetric storage capacities, respectively. However if we compare the pore properties of UiO MOFs (Table 1) and the deliverable capacity shown in Fig. 3, it can be realized that at optimal Qst, such hypothesis does not seem to be valid. For instance, at optimal Qst, both UiO-67 and UiO-68 exhibit the same level of volumetric (deliverable) storage capacity while the latter structure also exhibits a higher gravimetric storage capacity due to the larger surface area. It might be the case that there does not exist any simple single relation between the pore volume and the volumetric storage capacity, if the hydrogen adsorption properties are treated in terms of deliverable capacity. Furthermore it is evident from Fig. 3, that only UiO-68 could meet simultaneously the volumetric and gravimetric targets of 2010 at optimal Qst. In that case the pore properties UiO-68 that has a surface area of 4162 m2 g−1 and a pore volume of 1.7 cm3 g−1 can be taken as a reference while working towards a storage material that can deliver H2 that meets the gravimetric and volumetric targets of 2010. | ||
Fig. 3 Deliverable capacity from 120 to 1 bar vs. the average isosteric heat of adsorption (for chemisorption potentials: 2εLa/H2–7εLa/H2) in UiO MOFs. Qst corresponds to the pressure range 0–1 bar where the fluid–fluid interactions are negligible when compared to solid–fluid interactions. ( : UiO-66; : UiO-66; : UiO-67; : UiO-67; : UiO-68); (closed and open symbols correspond to gravimetric and volumetric storage capacity, respectively). : UiO-68); (closed and open symbols correspond to gravimetric and volumetric storage capacity, respectively). | ||
Conclusions
In order to meet the DOE targets for materials based hydrogen storage the starting material should contain a large surface area and pore volume, such as UiO-67 and UiO-68. Improving the isosteric heat of adsorption in materials similar to UiO-66 containing only ultra-micropores and low pore volume might not be a right strategy to reach the DOE goals (at the studied pressure conditions). Irrespective of the pore properties, Qst in general improves the storage capacity. On the other hand, the results from this work based on the deliverable capacity suggest that high gravimetric and volumetric storage capacities could be achieved only by proper tuning of pore properties towards high surface area, larger pore volume and optimum isosteric heat. At least for UiO MOFs, this corresponds to the pore properties of UiO-68 with an optimal Qst of 29 kJ mol−1.Acknowledgements
The partial support by the ATLAS-H2 FP7 Marie Curie project (PIAP-GA-251562) is gratefully acknowledged. Thanks to Prof. Randall Snurr for generously providing us the MUSIC code.References
- K. M. Thomas, Catal. Today, 2007, 120, 389–398 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 20, 9848–9851 RSC.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- D. A. Gomez and G. Sastre, Phys. Chem. Chem. Phys., 2011, 13, 16558–16568 RSC.
- U.S. Drive Partnership, Dep. o Energy, https//www1.eere.energy.gov/vehiclesandfuels/pdfs/program/hstt_roadmap_june2013.pdf, 2013.
- X. Zou, M.-H. Cha, S. Kim, M. C. Nguyen, G. Zhou, W. Duan and J. Ihm, Int. J. Hydrogen Energy, 2010, 35, 198–203 CrossRef CAS PubMed.
- K. Srinivasu and S. K. Ghosh, J. Phys. Chem. C, 2011, 115, 16984–16991 CAS.
- N. Park, K. Choi, J. Hwang, D. W. Kim, D. O. Kim and J. Ihm, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19893–19899 CrossRef CAS PubMed.
- M. Dixit, T. Adit Maark, K. Ghatak, R. Ahuja and S. Pal, J. Phys. Chem. C, 2012, 116, 17336–17342 CAS.
- Q. Yang, A. D. Wiersum, P. L. Llewellyn, V. Guillerm, C. Serre and G. Maurin, Chem. Commun., 2011, 47, 9603–9605 RSC.
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcajada, T. Devic, E. Magnier, F. Cuevas, G. Férey, P. L. Llewellyn and M. Latroche, Dalton Trans., 2011, 40, 4879–4881 RSC.
- S. Chavan, J. G. Vitillo, M. J. Uddin, F. Bonino, C. Lamberti, E. Groppo, K.-P. Lillerud and S. Bordiga, Chem. Mater., 2010, 22, 4602–4611 CrossRef CAS.
- Y.-S. Bae and R. Q. Snurr, Microporous Mesoporous Mater., 2010, 132, 300–303 CrossRef CAS PubMed.
- R. F. Cracknell, Phys. Chem. Chem. Phys., 2001, 3, 2091–2097 RSC.
- Q. Wang and J. K. Johnson, J. Chem. Phys., 1999, 110, 577 CrossRef CAS PubMed.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06295g |
| This journal is © The Royal Society of Chemistry 2014 |