DOI:
10.1039/C4RA06270A
(Paper)
RSC Adv., 2014,
4, 38405-38411
Paclitaxel prodrug nanoparticles combining chemical conjugation and physical entrapment for enhanced antitumor efficacy
Received
26th June 2014
, Accepted 13th August 2014
First published on 13th August 2014
Abstract
This study presents the combination of the prodrug approach with the self-assembly technique by using a poly(ethylene glycol)–paclitaxel (PTX) conjugate as the nanomicellar carrier for delivery of free PTX drugs. The conjugate PEG-PCPGE/PTX readily forms stable micelles in aqueous solution and is used to form an intracellular pool of PTX after the micelles are endocytosed, where encapsulated drug is first released, then destabilized polymer–PTX molecules liberate free PTX following cleavage in the intracellular environment. The use of the conjugate PTX as the core of the micelles provides an ideal affinity site for free PTX, in the mean time, high drug loading and time-programmable delivery are realized.
Introduction
Over the past few decades, a number of approaches have been proposed to achieve high drug loading, site-specific and time-programmable delivery of therapeutics, and to alleviate undesired side effects.1 Among these methods, polymeric micelles formed from amphiphilic block copolymers have been extensively investigated as drug delivery systems. As drug carriers, micelles have manifested several attractive features and advantages over other types of carrier, such as core–shell structure with the drug in the core and thus effectively protected, low toxicity in the human body, and prolonged circulation time in the blood.2 In addition, the passive accumulation of the micelles in a solid tumor is achieved by the enhanced permeability and retention (EPR) effect of the vascular endothelia at the tumor.3
However, in spite of the successes both in vitro and in vivo, burst drug release cannot be avoided for polymeric micelles because of their dynamic instability.4 To overcome burst release, it was proposed that drug molecules be chemically combined to a copolymer chain to form a polymer–drug conjugate, which may be considered as a “polymeric prodrug”, as the drug may lose its medical activity in its conjugate state and can restore its medicine activity after being released in its pristine form from the conjugate.5 This prodrug methodology has advantages in solubilization of the drug and tunability of the drug pharmacokinetics. The advancements in this field are associated with the work by Kazunori Kataoka,6 Leroux,7 and Ulbrich.8 Polymer carriers such as poly(ethylene glycol) (PEG),5,9 poly[N-(2-hydroxypropyl)methacrylamide] (PHPMA),8,10 poly(amino acids)11 are being studied intensively and have been successfully used in clinical research.
The combination of the prodrug approach with self-assembling technique by using the prodrug as the micellar carrier could result in co-existence of the encapsulated drug and the conjugated drug in the same micelle, like a sort of “combination therapy”, which could increase the drug content and improve the drug release kinetics has been proposed and investigated. For example, Shen et al. conjugate camptothecin (CPT) molecules to a very short oligomer chain of ethylene glycol (OEG), forming amphiphilic phospholipid-mimicking prodrugs. The prodrugs formed stable liposome-like nanocapsules with a CPT loading content as high as 58 wt% with no burst release in aqueous solution.1a Yang et al. reported polymeric micelle drug delivery by encapsulating free PTX in poly(L-γ-glutamylglutamine)–paclitaxel conjugate.12 However, the drug loading content was only 7 wt%. Lu et al. encapsulated free PTX into PEGylated-paclitaxel nanoparticles and the hydrophobic of the polymer was composed by PTX molecule and it may not form stable micelles and the drug loading content was less than 10 wt%.9c
Herein, we design functionalized poly(ethylene glycol)–paclitaxel conjugate prodrug as nanomicellar carrier for delivery of free PTX as shown in Scheme 1. Firstly, a hydrophilic diblock copolymer, poly(ethylene oxide)-block-poly(allyl glycidyl ether) (mPEG-b-PAGE), was synthesized. The allyl groups on the PAGE block were converted to carboxyl groups by the thiol–ene reaction with 3-mercapto-propanoic acid (MPA) and the carboxyl groups were used to conjugate anticancer drug PTX. The conjugate PEG-PCPGE/PTX readily forms stable micelles in aqueous solution and is used to encapsulate free PTX. After the micelles were endocytosed, the encapsulated drug was first released and then the conjugated one released after it cleavage from the polymer in intracellular environment.
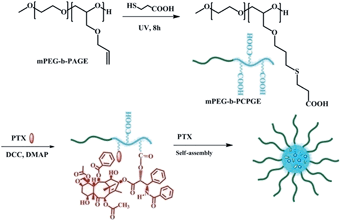 |
| | Scheme 1 Synthesis and self-assembly of PEG-PCPGE/PTX. | |
Synthesis of mPEG-b-PAGE
Firstly, block copolymer mPEG-b-PAGE was synthesized according to the literature as shown in Scheme 1.13 Secondly, in order to introduce carboxyl groups, MPA was used to react with the mPEG-b-PAGE by thio–ene reaction. Briefly, the mPEG-b-PAGE (1 g, 0.167 mmol) and MPA (0.58 mL, 6.67 mmol) were dissolved in 10 mL THF in a 100 mL round-bottom quartz flask, followed by degassing using N2 for 30 min to eliminate the dissolved oxygen. Then the mixture was stirred at room temperature under UV light (254 nm, 1.29 mW cm−2). After 8 h, the light source was turned off and the mixture was concentrated under vacuum. The residue was poured into large amount of cold diethyl ether to cause precipitation to give the final product mPEG-b-PCPGE (yield: 90%).
Synthesis of mPEG-b-PCPGE/PTX (P-PTX)
The antitumor drug PTX was conjugated to the copolymer mPEG-b-PCPGE via a DCC-catalyzed condensation reaction.14 Briefly, 0.2 g of mPEG-b-PCPGE was dissolved in 10 mL of dry CH2Cl2. Then 57 mg of PTX (66.8 mmol), 13.8 mg of dicyclohexylcarbodiimide (DCC, 66.8 mmol), and 8.2 mg of 4-dimethylaminopyridine (DMAP, 66.8 mmol) were added successively to the solution. The mixture was cooled to 0 °C and stirred under argon atmosphere for 24 h. Finally, dicyclohexylurea (DCU) formed was filtrated out and the filtrate was precipitated in a large amount of diethyl ether. The conjugate mPEG-b-PCPGE/PTX (abbreviated as “P-PTX” hereafter) was collected and dried in vacuo for 4 h (yield: 90%).
In order to trace the path of the conjugate in the cell, fluorescent dye Rhodamine B (Rho B) was conjugated to the end of the copolymer mPEG-b-PAGE by the similar condensation reaction above mentioned between the terminal hydroxyl group of mPEG-b-PAGE and the carboxyl group of Rho B. The obtained conjugate was abbreviated as P-Rho B. Hybrid micelles of P-PTX and P-RhoB were prepared by dissolving the mixture P-PTX and P-RhoB (weight ratio 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in 2 mL THF. Then the polymer solution was added dropwise to 10 mL of deionized water. The suspension was stirred for 10 h until no THF was detected. The RhoB labeled micelles was used for cellar uptake detection.
:1) in 2 mL THF. Then the polymer solution was added dropwise to 10 mL of deionized water. The suspension was stirred for 10 h until no THF was detected. The RhoB labeled micelles was used for cellar uptake detection.
Preparation of drug-loaded micelles
The obtained conjugate P-PTX was used as amphiphilic block copolymer for forming the core–shell structure to encapsulate hydrophobic drugs in the core part. The anticancer drug PTX was loaded into P-PTX micelles by solvent evaporation method. Briefly, PTX and P-PTX at specific weight ratios were dissolved in 4 mL THF. After 5 min of stirring, the solution was added dropwise to 10 mL of deionized water and stirred to evaporate organic solvent. Unincorporated PTX (precipitate) was removed by filtering with a syringe filter (pore size: 0.45 μm) and micelle powders (abbreviated as “M(PTX)”) were obtained by freeze-drying.
Physicochemical characterization
1H NMR spectra were measured in CDCl3 or DMSO-d6 at room temperature (20 ± 1 °C) by an AV-400 NMR spectrometer from Bruker. Size and size distribution of the micelles were determined by dynamic light scattering (DLS) with a vertically polarized He–Ne laser (DAWN EOS, Wyatt Technology, USA). The scattering angle was fixed at 90° and the measurement was carried out at 25 °C. The morphology of the micelles was measured by Transmission Electron Microscopy (TEM) performed on a JEOL JEM-1011 electron microscope operating at an acceleration voltage of 100 kV. To prepare specimens for TEM, a drop of micelle solution (1 mg mL−1) was deposited onto a copper grid with a carbon coating. The specimens were air-dried and measured at room temperature.
Drug encapsulation and loading efficiency
The PTX encapsulated in the micelles was measured by high-performance liquid chromatography (HPLC, Shimadzu, CBM-20A) with a UV-vis detector. A total of 5 mg drug-loaded micelle powder was dissolved in 2 mL of mobile phase (methanol–acetonitrile–water 32.5:32.5:35 v/v) and then was injected through a 20 μL sample loop. The elution rate was 1.0 mL min−1. Paclitaxel was detected at 227 nm. The retention time of paclitaxel was 7.5 min under these conditions.
The drug loading content (DLC) and drug loading efficiency (DLE) were calculated by the following equations:
| | |
DLC (%) = the free drug amount in the micelles/the amount of micelles
| (1) |
| | |
DLE (%) = the free drug amount in the micelles/the drug feeding
| (2) |
In vitro drug release
Release of PTX from the conjugate P-PTX and PTX-loaded mPEG-b-PCPGE/PTX micelles M(PTX) was performed in phosphate-buffered saline solution (PBS containing 0.5% Tween 80, 0.1 M, pH 7.4) at 37 °C. The solubility of PTX in PBS containing 0.5% Tween 80 is tested to be 0.042 mg mL−1. The initial concentration of PTX in micelles solution should not exceed this value very much to insure no precipitation in release medium during the test. Briefly, a total of 5 mg P-PTX or M(PTX) were dispersed in 2.5 mL of PBS and the dispersion was placed in a dialysis bag (MWCO = 3.5 kDa) that was incubated in 20 mL PBS containing 0.5% (w/v) Tween 80 at 37 °C with gentle shaking. 1 mL of release medium was withdrawn from the incubation medium at specified time intervals and measured by HPLC. After sampling, equal volume of fresh buffered solution was immediately added into the release medium. The PTX released from the micelles was expressed as the cumulative percentage of the PTX outside the dialysis bag to the total PTX in the micelles, expressed as the mean from triplicate samples.
MTT (3-(4,5-dmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay
Cell lines and cell culture
EMT6 (mouse mammary tumor cell) and MCF7 (human mammary tumor) cell lines were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China. They were grown in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS, GIBCO), and the culture medium was replaced once very day.
Cytotoxicity test
EMT6 and MCF7 cells harvested in a logarithmic growth phase were seeded in 96-well plates at a density of 105 cells per well and incubated in DMEM for 24 h. The medium was then replaced by blank polymer mPEG-b-PCPGE (BP), P-PTX, PTX, and M(PTX), at a final polymer concentration from 0.001 to 10 μg mL−1 for blank polymer mPEG-b-PCPGE and an equivalent drug concentration from 0.001 to 10 μg mL−1 for free drug and drug loaded micelles. The incubation was continued for 48 h. Then, 20 μL of MTT solution in PBS with the concentration of 5 mg mL−1 was added and the plates were incubated for another 4 h at 37 °C, followed by removal of the culture medium containing MTT and addition of 150 μL of DMSO to each well to dissolve the formazan crystals formed. Finally, the plates were shaken for 5 min, and the absorbance of formazan product was measured at 490 nm by a microplate reader.
Cellular uptake
Cellular uptake by MCF-7 human ovarian cancer cells was examined using confocal laser scanning microscope (CLSM). MCF-7 cells were seeded in 6-well culture plates (a clean cover slip was put in each well) at a density of 5 × 104 cells per well and allowed to adhere for 24 h. RhoB labeled micelles were added. After incubation 0.5 h, 1 h and 4 h at 37 °C, the supernatant was carefully removed and the cells were washed three times with ice-cold PBS. Subsequently, the cells were fixed with 800 μL of 4% formaldehyde each well for 10 min at room temperature and washed twice with ice-cold PBS again. Samples were examined by CLSM using a Zeiss LSM 510 (Zurich, Switzerland) with a confocal plane of 300 nm. RhoB was excited at 488 nm with emissions at 560 nm.
Antitumor efficacy
Balbc female mice were obtained from Jilin University, China (56–84 days, 15–20 g) and maintained under required conditions. Use of them for this study was approved by the Animal Ethics Committee of Jilin University. To develop the tumor xenografts, EMT 6 cells were injected into the lateral aspect of the anterior limb of the mice (5 × 106 cells in 0.1 mL PBS). After the tumor volume reached 20–50 mm,3 the hair of the mice was removed with a sodium sulfide solution (80 g L−1 in 30 vol% aqueous alcohol).
Thirty-two mice bearing EMT6 tumor nodules were randomly divided into four groups: (1) normal saline (blank control), (2) PTX, (3) P-PTX and (4) M(PTX) groups. Before injection, all the mice were marked and weighed, and the length and width of the tumor were measured as the initial size on day 1. The day of starting injection was designated as day 1. For groups (3) and (4), an equivalent PTX dose of 40 mg kg−1 per body was intravenously injected via tail vein on day 1, day 3 and day 5. And for PTX, the dose was 15 mg kg−1. For group (1), the mice were injected with an equivalent volume of normal saline. The tumor size was measured every other day, and the tumor volume (V) was calculated by the formula V = ab2/2, where a and b were the length and width of tumor, respectively. The mouse survival rate in each group was recorded daily.
Statistics
All experiments were performed at least three times and all results are expressed as mean ± (standard deviation). Student's t-test was used to demonstrate statistical significance (P < 0.05).
Results and discussion
Synthesis of mPEG-b-PCPGE
Poly(ethylene glycol) (PEG) is the most commonly used hydrophilic polymer for drug delivery owning to its unique properties, such as: low immunogenicity and toxicity, high solubility in water and in many organic solvents, and approval by FDA for human use. The aim of this study was to design a drug delivery system with high drug loading and time-controlled release based on functionalized PEG by combination prodrug approach and self-assembling technique.
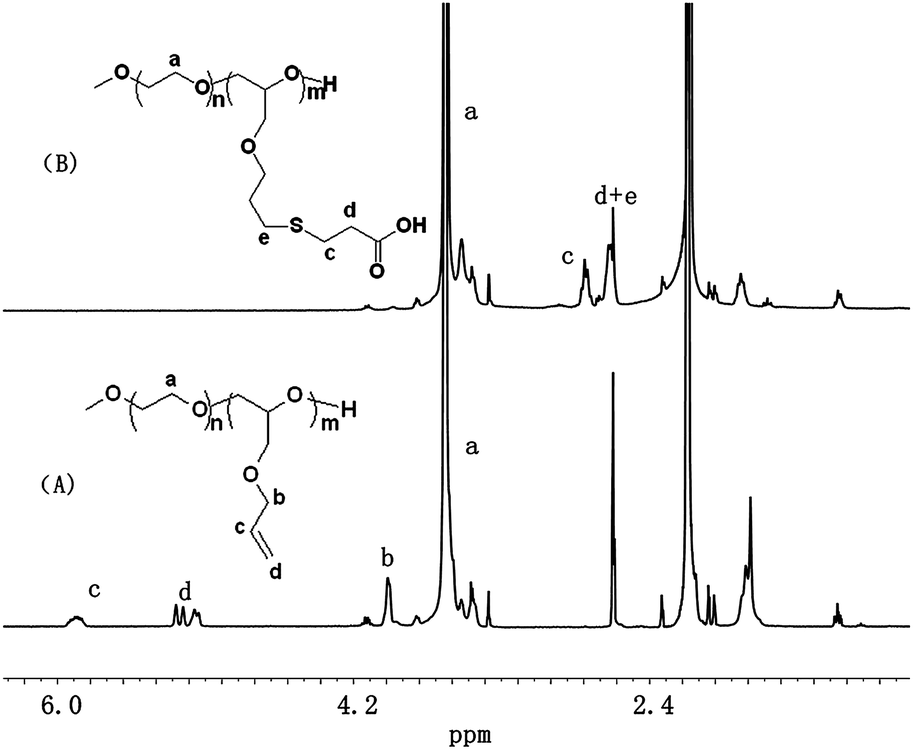
Firstly, the copolymer mPEG-b-PAGE was synthesized by anionic ring-opening polymerization of allyl glycidyl ether (AGE) using PEG5000 as the initiator according to the ref. 13. Two polymers were synthesized by adjusting the feed ratio of macroinitiator and polymerization monomers allyl glycidyl ether (AGE). And the polymerization degrees of PAGE block were designed to be 5 and 10, respectively. The typical 1H NMR spectra of mPEG-b-PAGE in CDCl3 were shown in Fig. 1. As can be seen in Fig. 1, mPEG-b-PAGE displayed the 1H NMR signals of mPEG ((a) at 3.65 ppm) and PAGE ((b) at 4.0 ppm, (c) at 5.9 ppm and (d) at 5.3 ppm). The polymerization degrees of AGE in the copolymer were calculated to be 5 and 9 from the integral area ratios of –CH2CH2O (mPEG at 3.65 ppm) to CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2 ((c) at 5.9 ppm). Then the allyl groups on PAGE block were converted to carboxyl groups via the thio–ene reaction with MPA to obtain pendant carboxyl functionalized diblock copolymer poly(ethylene glycol)-block-poly((2-carboxy-ethylsulfanyl)-propyl glycidy ether) (PEG-PCPGE). After the radical mediated thiol–ene addition reaction between the allyl groups and MPA, the signals of the allyl groups disappeared along with appearance of several CH2 groups ((c) at 2.76 ppm and (d) at 2.61 ppm Fig. 1(B)), which were the proton of methylene adjacent to carboxylic groups. The obtained copolymers were also characterized by GPC and all the five copolymers show narrow molecular weight distributions (MDI = 1.03–1.1, Table 1). The molecular weights determined by GPC were in agreement with those determined by 1H NMR (Table 1). All these results suggest that the polymerization proceeded in a controlled manner to yield well-defined copolymers.
CH–CH2 ((c) at 5.9 ppm). Then the allyl groups on PAGE block were converted to carboxyl groups via the thio–ene reaction with MPA to obtain pendant carboxyl functionalized diblock copolymer poly(ethylene glycol)-block-poly((2-carboxy-ethylsulfanyl)-propyl glycidy ether) (PEG-PCPGE). After the radical mediated thiol–ene addition reaction between the allyl groups and MPA, the signals of the allyl groups disappeared along with appearance of several CH2 groups ((c) at 2.76 ppm and (d) at 2.61 ppm Fig. 1(B)), which were the proton of methylene adjacent to carboxylic groups. The obtained copolymers were also characterized by GPC and all the five copolymers show narrow molecular weight distributions (MDI = 1.03–1.1, Table 1). The molecular weights determined by GPC were in agreement with those determined by 1H NMR (Table 1). All these results suggest that the polymerization proceeded in a controlled manner to yield well-defined copolymers.
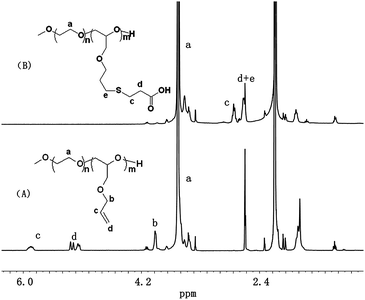 |
| | Fig. 1 1H NMR spectra of mPEG-b-PAGE (A) and mPEG-b-PCPGE (B) in CDCl3. | |
Table 1 Characterization of block copolymers
| Copolymers |
Mna (kg mol−1) |
Mnb (kg mol−1) |
PDIb |
| Determined by 1H NMR. Determined by GPC. |
| mPEG113 |
5.0 |
5.1 |
1.03 |
| mPEG113-PAGE5 |
5.5 |
5.7 |
1.08 |
| mPEG113-PAGE9 |
6.0 |
6.5 |
1.06 |
| mPEG113-PAGE5 |
6.1 |
6.7 |
1.08 |
| mPEG113-PAGE9 |
7.2 |
8.0 |
1.1 |
Conjugate with PTX
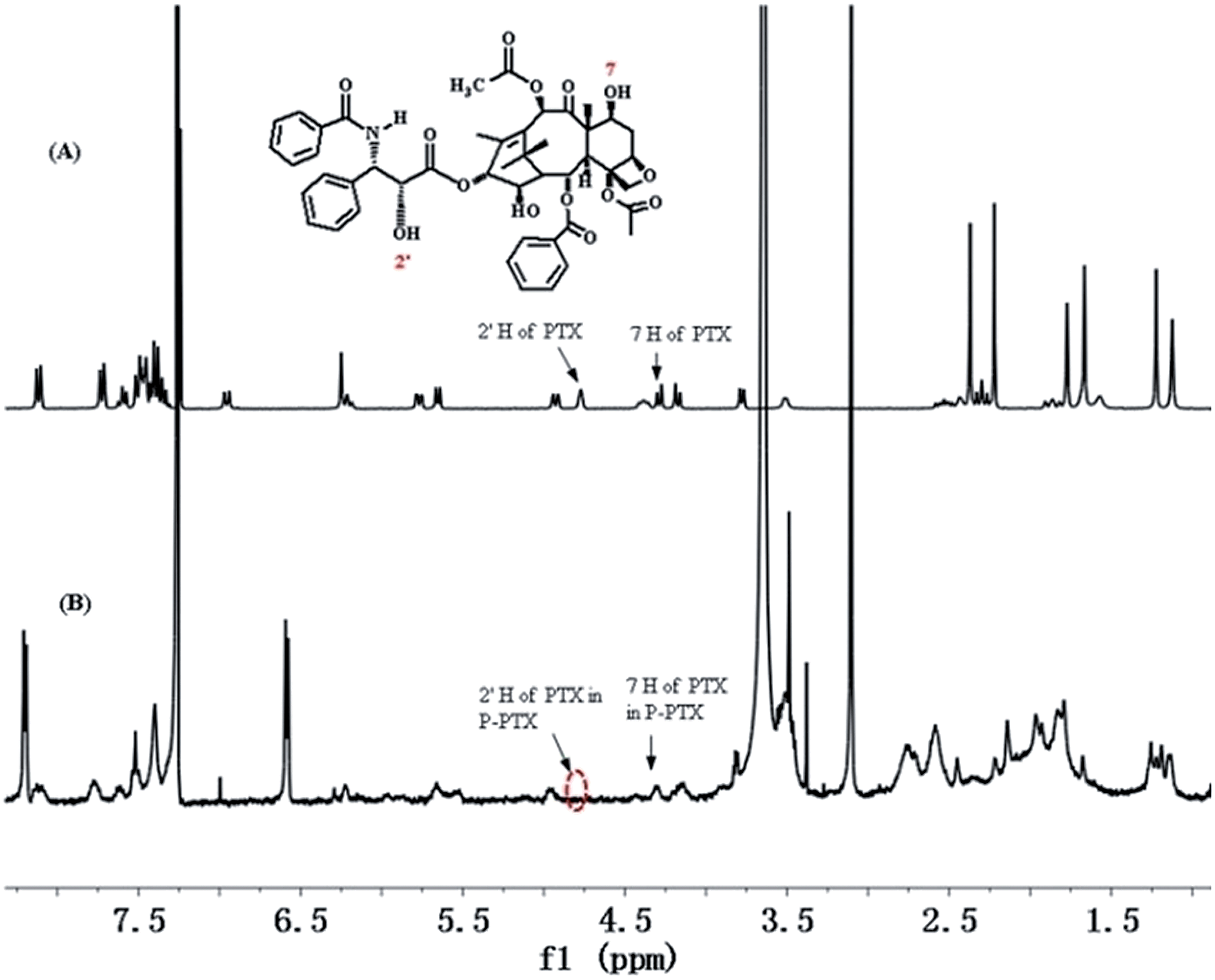
The hydroxyl group in the 2′ position of PTX is known to be fairly reactive and its chemical derivatization has been largely explored, including the formation of ester prodrugs.15 Consistently, reaction of PTX with pendant carboxyl groups of mPEG-b-PCPGE in dichloromethane by DCC/DMAP-catalyzed esterification reaction readily afforded mPEG-b-PCPGE/PTX conjugate. The copolymer mPEG113-PCPGE9 was selected and used for drug conjugation and different PTX molecules were designed to conjugate to each polymer chain by adjusting the molar ratio of PTX to COOH. PTX:COOH ratios was defined to be 2:9, 3:9 and 5:9, respectively, and which means there were 2, 3, and 5 PTX molecular conjugated to each polymer chain, respectively. HPLC was used to monitor the reaction process to ensure complete conversion of all the PTX added and the structure of the resulting conjugate PEG-PCPGE/PTX was confirmed by 1H NMR analysis (Fig. 2). In the 1H NMR spectrum of PEG-PCPGE/PTX, the characteristic resonance of 7-CH at 4.45 ppm remained, while the resonance of 2′-CH proton was shifted from 4.77 ppm to 5.55 ppm. The content of PTX in the conjugate was calculated according to its 1H NMR spectrum to be 19 wt%, 26.4 wt%, and 33 wt%, respectively (Table 2).
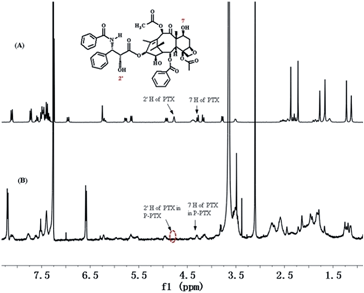 |
| | Fig. 2 1H NMR spectra of PTX (A) and P-PTX 2 (B) in CDCl3. | |
Table 2 Characterization of polymer–paclitaxel conjugates (P-PTX)
| Sample |
PTX:COOH |
PTX content |
Dha (nm) |
PDIa |
| Determined by DLS. |
| P-PTX 1 |
2:9 |
19 wt% |
35 |
0.266 |
| P-PTX 2 |
3:9 |
26.4 wt% |
60 |
0.289 |
| P-PTX 3 |
5:9 |
33 wt% |
260 |
0.299 |
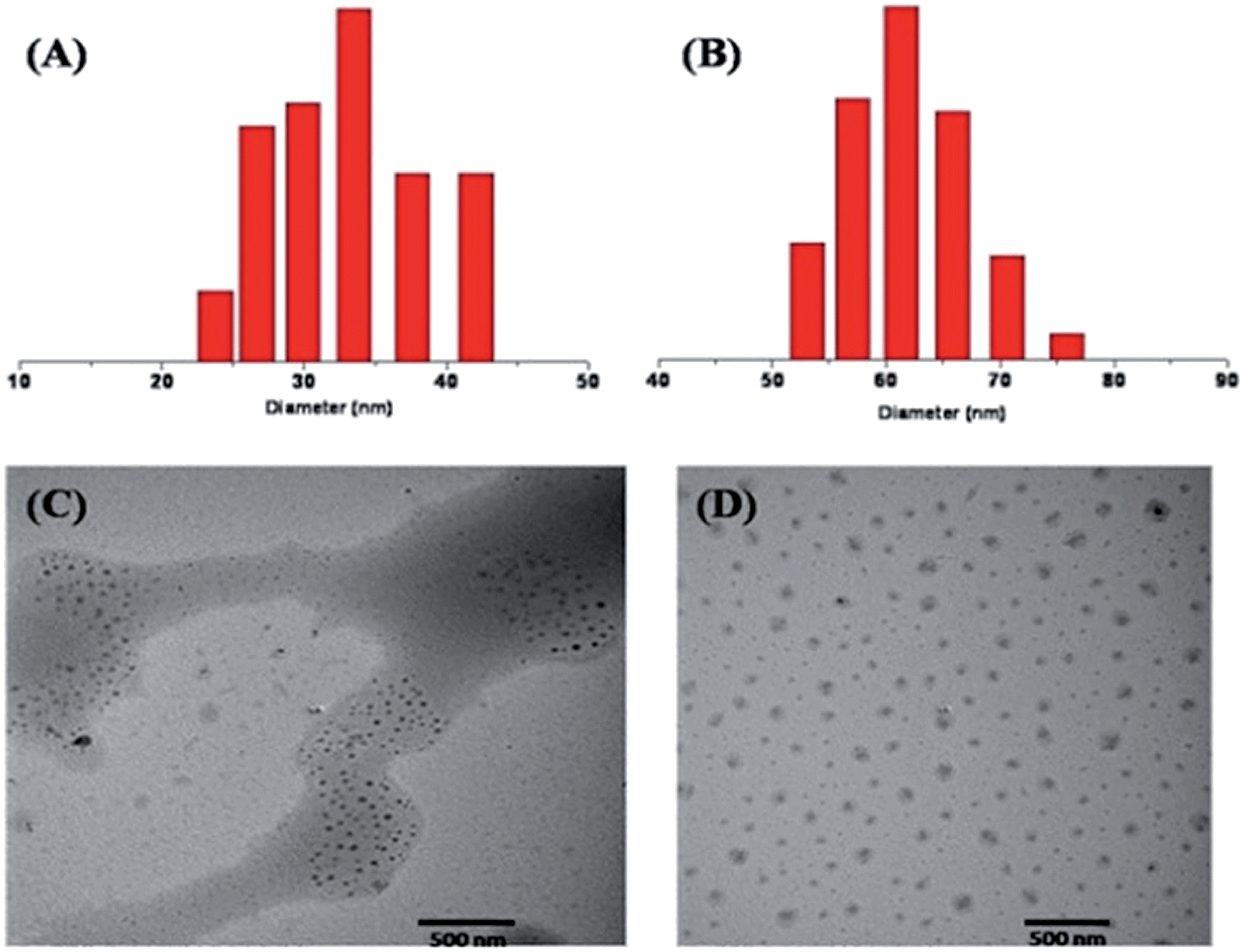
The obtained prodrug P-PTX1 and P-PTX2 readily form micelles in aqueous solution and their hydrodynamic diameter were around 35 nm and 60 nm at 0.5 mg mL−1 in water, respectively. The typical size and size distribution of P-PTX1 and P-PTX2 measured by dynamic light scattering (DLS) (Fig. 3(A) and (B)) and transmission electron microscopy (TEM) (Fig. 3(C) and (D)) analysis was show in Fig. 3. For P-PTX3, there were obvious aggregates using the same preparation method as the first two conjugates. The sudden increased diameter of its corresponding micelles may due to its aggregation. The CMC of P-PTX1 and P-PTX2 were determined by using a pyrene probe according to the literature and the value is 0.06 and 0.01 g L−1, respectively.
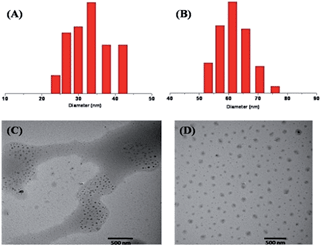 |
| | Fig. 3 Particle size distribution of P-PTX1 (A) and P-PTX2 (B); TEM image of P-PTX1 (C) and P-PTX2 (D). | |
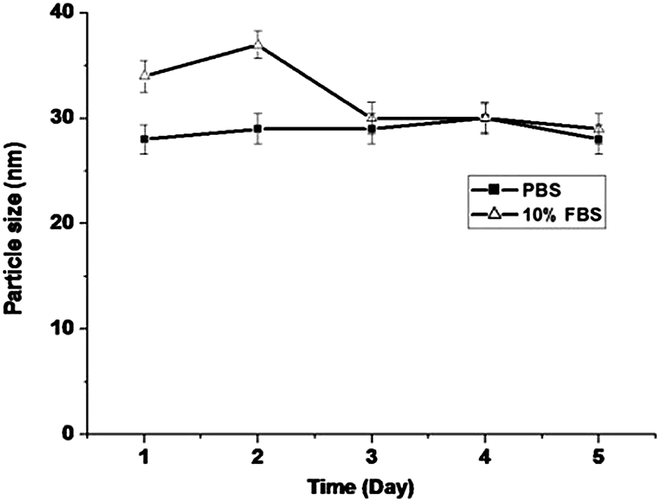
The stability of P-PTX micelles was examined by measuring the average size as a function of standing time in pH 7.4 PBS and in pH 7.4 PBS containing 10% FBS (Fetal Bovine Serum). As shown in Fig. 4, the average size of P-PTX micelles remains almost unchanged within five days in both systems.
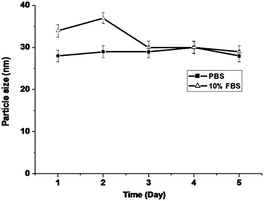 |
| | Fig. 4 P-PTX micelle stability measured by dynamic light scattering (n = 3) in pH 7.4 PBS and 10% FBS solution. Incubation temperature: 37 °C. | |
Drug encapsulation
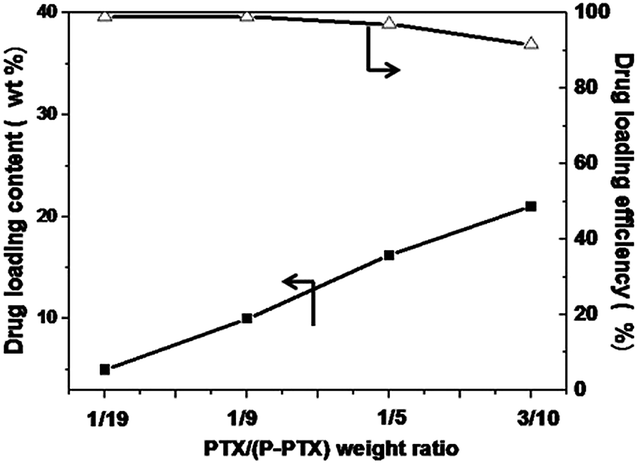
The P-PTX1 micelle was further used to encapsulate hydrophobic anticancer drugs, such as PTX. Micelles with different drug/polymer ratios were prepared. Almost all of the added PTX was entrapped into the micelles formed to achieve drug content from 5 wt% to 16.7 wt% and drug loading efficiency of almost 100% (Fig. 5). Such high loading efficiency was attributed to the existence of the PTX moieties in the P-PTX carrier. There is no doubt that these PTX moieties have best affinity with the free PTX added. The highest drug loading content was 21 wt% with a loading efficiency of 91%. Keeping in mind that the P-PTX itself contains about 19 wt% of PTX, the total PTX content would be as high as 36 wt%.
 |
| | Fig. 5 PTX loading content and drug loading efficiency at different feeding weight ratios of drug to polymer. | |
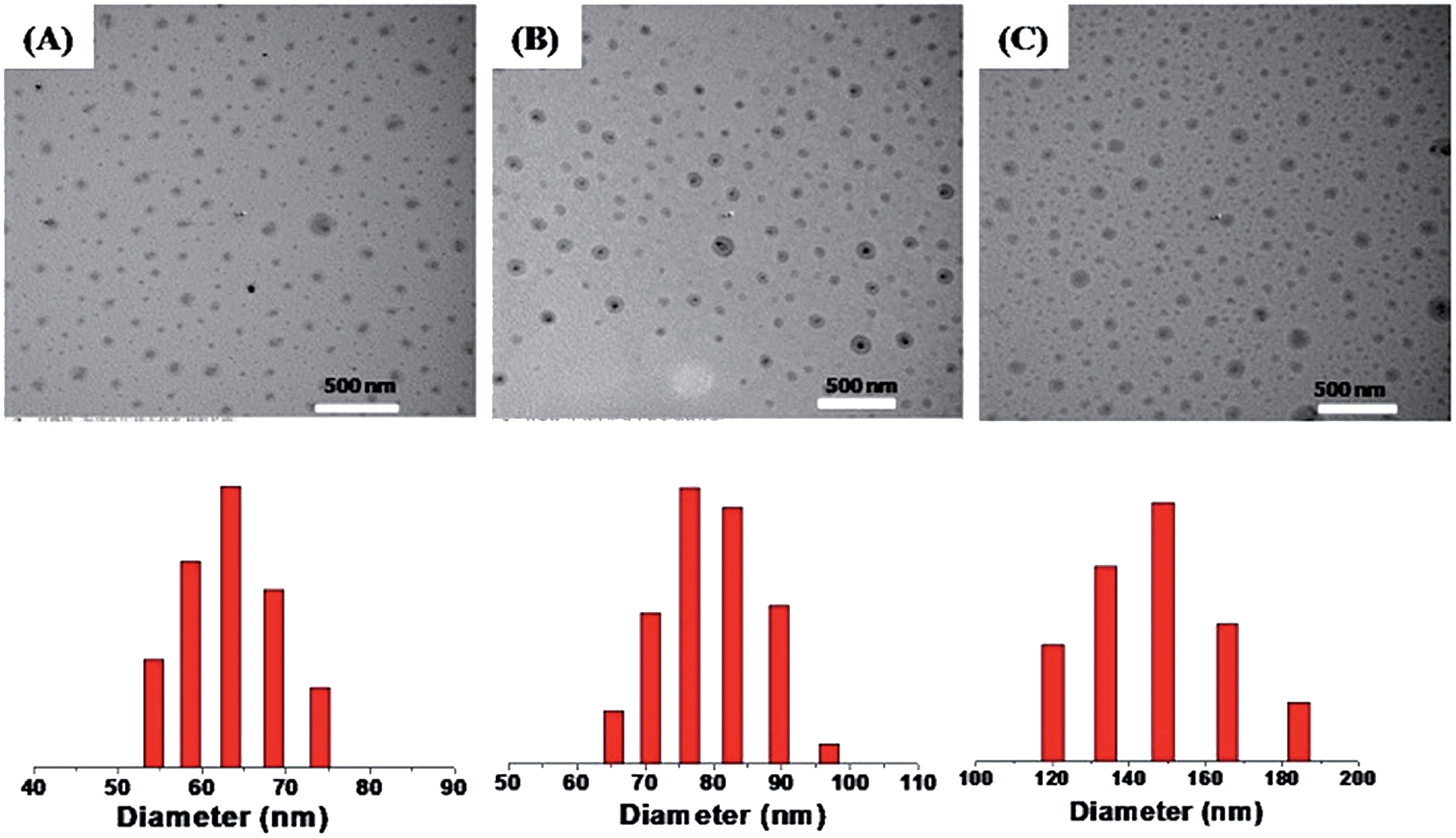
The size of drug loaded micelles was also characterized by DLS and TEM (Fig. 6), and it increased with the increase in drug content. The average diameters were 65 nm, 75 nm and 150 nm for PTX loading content of 10 wt%, 16.1 wt% and 21 wt%, respectively, as measured by DLS (Fig. 6).
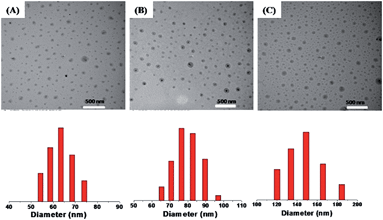 |
| | Fig. 6 TEM images (upper) and DLS plots (lower) of PTX loaded micelles with different loading contents: 10 wt% (A), 16.7 wt% (B) and 21 wt% (C). | |
In vitro release of PTX from M(PTX)
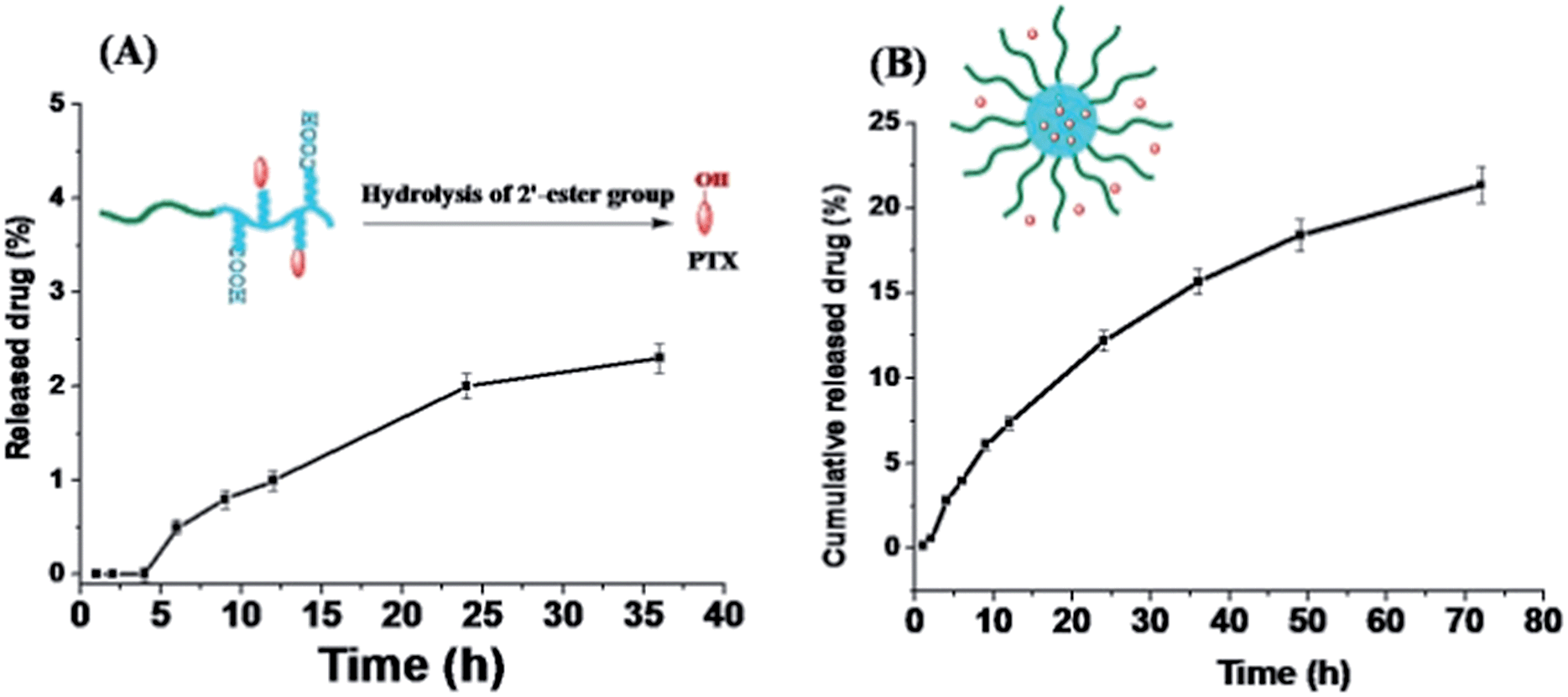
The PTX release profile of P-PTX and M(PTX) were examined by a dialysis method. According the chemical structure of the conjugate P-PTX, there is only one type of cleavable group in the conjugate, the 2′-ester group as shown in Fig. 7(A). Quantitative analysis of the release of PTX moieties from the conjugate was conducted by HPLC analysis of the buffer solution of P-PTX incubated under a physiological pH 7.4 at 37 °C. As illustrated in Fig. 7(A), the maximum PTX release percent attainable after a period of 36 h in PBS (pH = 7.4) was less than 5%. In line with the stability of PTX micelle confirmed by DLS measurement as shown in Fig. 4, the ester group between the polymer carrier and PTX was stable under this condition. And for the PTX encapsulated micelles M(PTX), PTX moieties were gradually released from micelles without burst effect (Fig. 7(B)). Taking into account that almost no conjugated PTX was released under this condition (Fig. 7(B)), the detected PTX was mainly the encapsulated one.
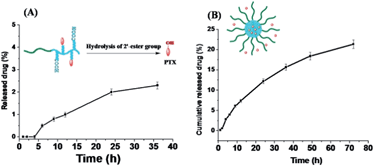 |
| | Fig. 7 Drug release profile of P-PTX (A) and M(PTX) (B) in phosphate buffer (pH = 7.4, 0.1 M) containing 0.5% w/v Tween 80 at 37 °C (data represents mean ± SD (n = 3). | |
In vitro cytotoxicity
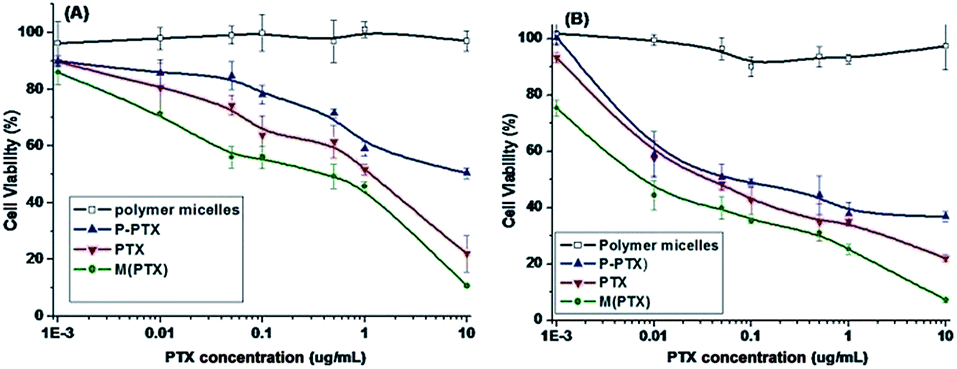
The cytotoxicities of blank polymer mPEG-b-PCPGE (BP), pure PTX, and prodrug P-PTX against EMT-6 and MCF-7 cells determined by MTT assay were illustrated in Fig. 8. The block copolymer mPEG-b-PCPGE was found relatively nontoxic to the cells at concentrations of 0.001 to 10 μg mL−1, indicating excellent biocompatibility of it. At the drug concentration range examined, the prodrug P-PTX showed comparable toxicity to small molecular PTX, indicated the drug molecules were released from the conjugate under intracellular environment. And free PTX containing micelles M(PTX) has the best anticancer activity.
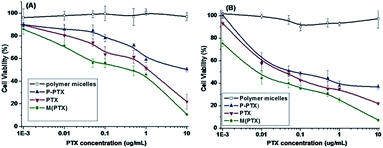 |
| | Fig. 8 In vitro cytotoxicity of mPEG-PCPAGE, P-PTX1, PTX and M(PTX) against EMT6 (A) and MCF-7 (B) cells for 48 h. | |
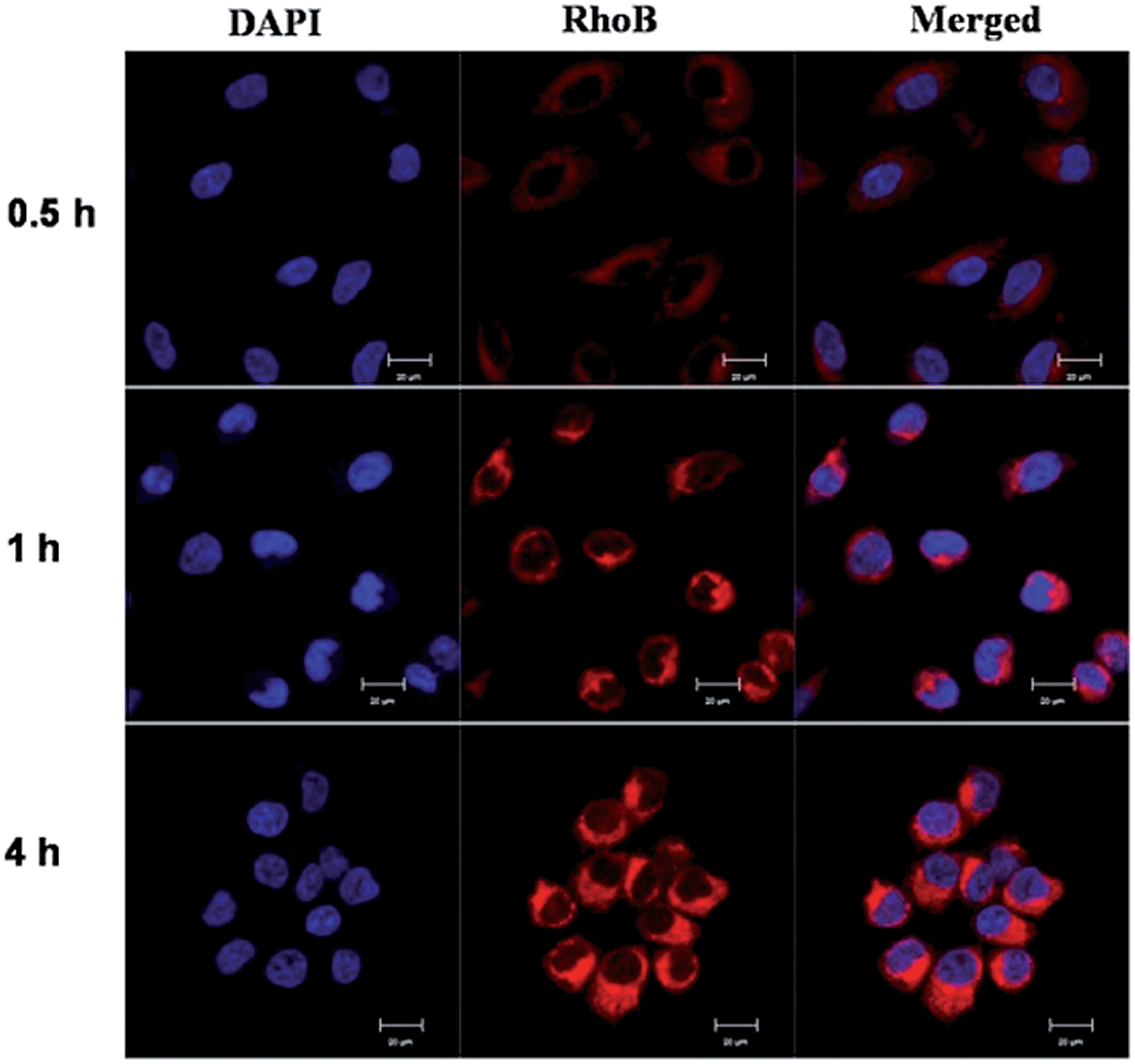
Cellular uptake. In order to visualize the conjugate micelles taken by the cells, MCF-7 cells were incubated with micelles co-assembled form Rhodamine B labeled copolymer P-RhoB and paclitaxel conjugated copolymer P-PTX for 0.5 h, 1 h and 4 h, respectively, and examined by confocal laser scanning microscopy (CLSM) (Fig. 9). The cellular nuclei were selectively stained with DAPI (blue). As shown in Fig. 9, after 0.5 h incubation, red fluorescence was observed in the cell plasma, indicating that micelles were taken up by the cell. And the red fluorescence in the cells increased obviously along with the incubation time at 1 h and 4 h. This suggested that more and more micelles were internalized and the rate of endocytosis of the micelles was efficiently.
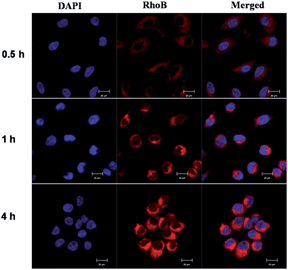 |
| | Fig. 9 Confocal laser scanning microscopy images of RhoB labeled P-PTX micelles after incubation with MCF-7 cells for 0.5 h, 1 h and 4 h, respectively. All pictures show the nuclei (blue), drug (RhoB) and merged images. Scale bars represent 20 μm in all images. | |
In vivo antitumor efficacy
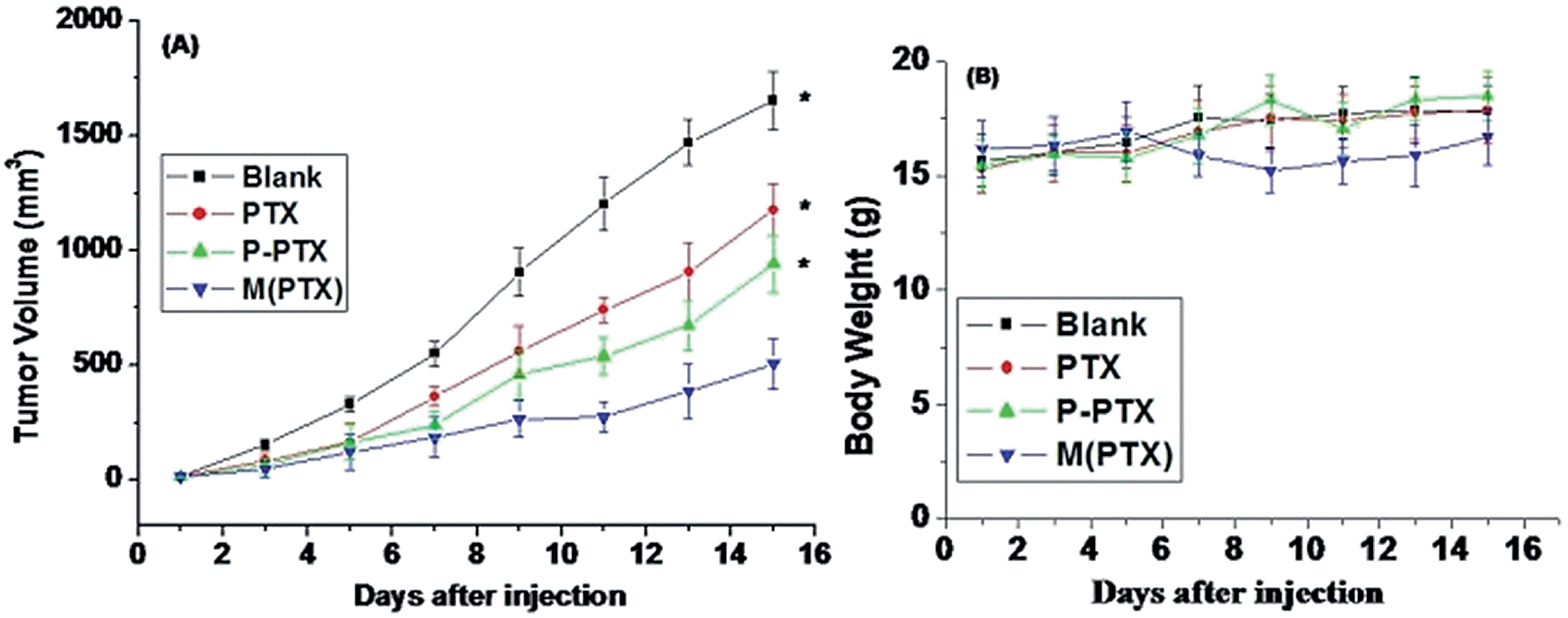
In order to study the in vivo antitumor effect of paclitaxel conjugated and entrapment micelles P-PTX and M(PTX), mouse breast xenograft models of EMT6 were established by injection of EMT6 cells into the lateral aspect of the anterior limb of balbc mice. When the tumor grew to a size of 10–50 mm3, 5 days after inoculation of the cancer cells, the mice were randomly divided into four groups with 8 mice in each group. The dose of free PTX per injection was set as 15 mg kg−1 because the mice will die by increasing the dose. And the dose of conjugate drug P-PTX and M(PTX) were 40 mg kg−1 per injection, all these three groups were given three times (on day 1, 3 and 5) via tail iv injection, and the tumor sizes were measured every other day. Fig. 10 shows the tumor size (A) and mice body weight change (B) as a function of time. It was notable that the administration of P-PTX was more efficacious in tumor suppression compared with free PTX and M(PTX) has the best antitumor efficacy. The mice body weight did not change much and all mice were alive for all these four groups.
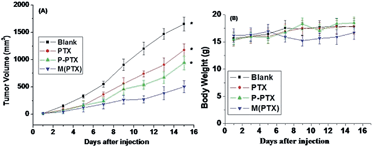 |
| | Fig. 10 (A) Relative tumor sizes of mouse models bearing EMT6 cancers as a function of time after drug administration. Bars indicate standard deviations. (B) Body weight change of mice bearing EMT6 cancers.*P < 0.05 vs. control. Results are the mean ± SD of eight independent experiments. | |
Conclusions
In summary, we have demonstrated the possibility of using drug molecules (PTX) as components of a polymer carrier to encapsulate small molecular drugs. The polymer–PTX conjugate was successfully prepared by linking PTX to the pendant carboxyl groups of mPEG-b-PCPGE and hydrophobic drugs PTX was effectively encapsulated into the polymer–PTX micelles with a drug loading capacity up to 21 wt% and with a drug loading efficiency of 91%. In addition, there was chemically combined PTX of 19 wt%. Accordingly, the non-drug component only took 63 wt%. The in vitro evaluation showed that this combination method substantially increases the drug loading content and drug loading efficiency, minimizes use of inactive materials, effectively adjusts drug release kinetics and helps realize co-loading of different drugs for combination chemotherapy. In vitro and in vivo antitumor experiments show that these micelles have better antitumor efficacy than free drug molecules.
Notes and references
-
(a) Y. Shen, E. Jin, B. Zhang, C. j. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259 CrossRef CAS PubMed;
(b) Y. Tian and S. Mao, Expert Opin. Drug Delivery, 2012, 9, 687 CrossRef CAS PubMed.
- X. Hu and X. Jing, Expert Opin. Drug Del., 2009, 6, 1079 CrossRef CAS PubMed.
- K. Ogawara, Y. Yoshizawa, K. Un, T. Araki, T. Kimura and K. Higaki, Biol. Pharm. Bull., 2013, 36, 698 CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37 CrossRef PubMed.
- X. Zhang, J. Lu, Y. Huang, W. Zhao, Y. Chen, J. Li, X. Gao, R. Venkataramanan, M. Sun, D. B. Stolz, L. Zhang and S. Li, Bioconjugate Chem., 2013, 24, 464 CrossRef CAS PubMed.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92, 1240 CrossRef CAS PubMed.
- G. Fuhrmann, M. A. Gauthier and J.-C. Leroux, Chimia, 2013, 67(9), 685 CrossRef.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2010, 62, 150 CrossRef CAS PubMed.
-
(a) F. M. Veronese and A. Mero, Biodrugs, 2008, 22(5), 315 CrossRef CAS PubMed;
(b) J. Lu, Y. Huang, W. Zhao, R. T. Marquez, X. Meng, J. Li, X. Gao, R. Venkataramanan, Z. Wang and S. Li, Biomaterials, 2013, 34(5), 1591 CrossRef CAS PubMed;
(c) J. Lu, X. Chuan, H. Zhang, W. Dai, X. Wang, X. Wang and Q. Zhang, Int. J. Pharm., 2014, 471(1–2), 525 CrossRef CAS PubMed.
-
(a) F. Greco and M. J. Vicent, Adv. Drug Delivery Rev., 2009, 61, 1203 CrossRef CAS PubMed;
(b) Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS PubMed.
-
(a) A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169 CrossRef CAS;
(b) Y. Bae and K. Kataoka, Adv. Drug Delivery Rev., 2009, 61, 768 CrossRef CAS PubMed;
(c) J. Guo, Y. Wei, D. Zhou, P. Cai, X. Jing, X. S. Chen and Y. Huang, Biomacromolecules, 2011, 12(3), 737 CrossRef CAS PubMed.
- D. Yang, S. Van, X. Jiang and L. Yu, Int. J. Nanomed., 2011, 6, 85 CAS.
- R. Wang, X. Hu, J. Yue, W. Zhang, L. Cai, Z. Xie, Y. Huang and X. Jing, J. Mater. Chem. B, 2013, 1, 293 RSC.
- X. F. Zhang, Y. X. Li, X. S. Chen, X. H. Wang, X. Y. Xu, Q. Z. Liang, J. L. Hu and X. B. Jing, Biomaterials, 2005, 26, 2121 CrossRef CAS PubMed.
- C. Clementi, K. Miller, A. Mero, R. Satchi-Fainaro and G. Pasut, Mol. Pharmaceutics, 2011, 8, 1063 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.