DOI:
10.1039/C4RA06251E
(Paper)
RSC Adv., 2014,
4, 35639-35648
Iridium(III) catalyzed trifluoroacetoxylation of aromatic hydrocarbons†
Received
11th March 2014
, Accepted 18th July 2014
First published on 18th July 2014
Abstract
A tridentate, NNC-tb (where NNC-tb = 2-(pyridin-2-yl)benzo[h]quinoline) ligated IrIII complex (NNC-tb)Ir(Ph)(4-MePy)(TFA), 11 along with analogues are very active for CH activation as evidenced by rapid catalytic H/D exchange between benzene and trifluoroacetic acid – d1 (DTFA). The complexes were examined with a variety of oxidants for the catalytic conversion of benzene to phenyltrifluoroacetate. Herein, the synthesis and characterization of (NNC-tb)Ir complexes is described along with the reactivity of these complexes towards arenes and alkanes.
Introduction
Catalytic CH activation followed by oxy-functionalization of hydrocarbons to generate alcohols using strong electrophiles has proven a viable strategy for hydrocarbon oxidation (Scheme 1).1–7 Systems based on soft, oxidizing cations such as HgII,8–10 PtII,11–22 PdII,23–27 and AuI/AuIII28,29 generate oxy-functionalized products from methane at product concentrations >1 M. However, the most active systems utilize concentrated, oxidizing acids and suffer from catalyst poisoning due to the generation of product and H2O that bind to the strongly electrophilic center.30 This inhibition has rendered these systems impractical and provided the motivation for the development of new systems that do not suffer from such inhibition.31
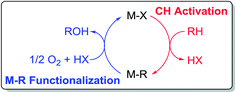 |
| | Scheme 1 General catalytic cycle for CH activation/M–R functionalization of hydrocarbons. | |
The use of less electrophilic, less oxidizing cations such as those based on IrIII or RhIII has been under exploration, as these species do not show sensitivities to water or product, and reactions can be carried out in non-oxidizing, relatively inert solvents such as CF3CO2H (HTFA).32–35 Indeed, several reports have now shown that these weakly electrophilic cations can undergo CH activation with hydrocarbons in carboxylic acid solvents to generate M–R intermediates.36–38 However, efficient catalysts have not been developed with these systems likely due to high energy barriers for oxy-functionalization of the M–R intermediate that is required for a complete catalytic cycle, Scheme 1.39–42
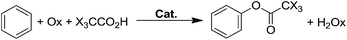
Recently, several groups have published examples of homogeneous catalyzed oxidations of aromatics with transition metal catalysts proposed to proceed via a CH activation mechanism (eqn (1)). For example, Sanford reported a Pd acetate complex that catalyzes H/D exchange of benzene and leads to the phenyl-ester when the system is treated with PhI(OAc)2 as the oxidant.43 Yu and coworkers reported several examples of the direct functionalization of aromatics with O2 also using Pd catalysts.44–46 An important thrust of our work has been the design of new homogeneous catalysts for the CH activation of alkanes and arenes. We recently reported IrIII complexes bearing the tBu-NNC ligand (tBu-NNC = 6-phenyl-4,4′-di-tert-butyl-2,2′-bipyridine) capable of undergoing CH activation with strong CH bonds such as methane and benzene.47–50 It was shown that (tBu-NNC)Ir(Et)(C2H4)(TFA), 1, in the presence of an oxidant, such as NaIO4, gave ∼6 turnovers (TON) of methyltrifluoroacetate after 3 h at 180 °C (Scheme 2). However, after 3 h, further H/D exchange or oxidation products were not observed. This is likely due to deactivation of the catalyst via pooling at higher oxidation states (e.g. Ir(IV or V)) that are likely not capable of undergoing CH activation.12
| |
 | (1) |
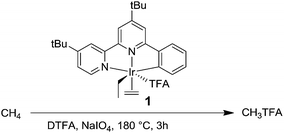 |
| | Scheme 2 Oxidation of methane to MeTFA by 1 in HTFA. | |
Another key issue with the (tBu-NNC)Ir system was the apparent lack of thermal stability of the (tBu-NNC)Ir systems above 100 °C that likely limited the efficiency of the system. Studies suggested that cleavage of the cyclometalated Ir–carbon bond, leading to loss/oxidation of the ligand or metal center, may have contributed to the complexes' low yield relative to the observed rates of H/D exchange and the observed loss of activity in extended run times. To address this, we investigated the use of new ligand designs to improve the stability of the catalytic system to the oxidizing and acidic reaction conditions. We also explored coupling the CH activation reaction to a functionalization reaction with potentially practical oxidants to generate phenol from benzene.
Results and discussion
In an effort to design a more robust catalytic system, similar to the originally reported NNC system, we explored related iridium complexes with a geometrically restricted or “tied back” NNC motif. In the original report of the Ir complexes and subsequent work, it was discovered that the cyclometalled ring underwent decyclometallation as observed by incorporation of deuterium selectively in the ortho position of the phenyl ring.50 We believe this loss of cyclometalation may be a root cause of the lack of thermal stability observed in these complexes. We postulated that fusing the phenyl ring to one of the pyridines should prevent the decyclometalation of the phenyl ring (Fig. 1) as it would require loss of the chelating ligand. This led to the synthesis and study of an (NNC-tb) (NNC-tb = 2-(pyridin-2-yl)benzo[h]quinoline) ligated Ir system. The expectation was that a fused benzoquinoline motif should restrict the degrees of freedom of the cyclometalated phenyl group and lead to higher barriers for decyclometallation and higher thermal stability.
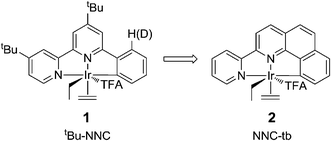 |
| | Fig. 1 Parent NNC ligand system (tBu-NNC) compared to the “tied back” motif (NNC-tb). | |
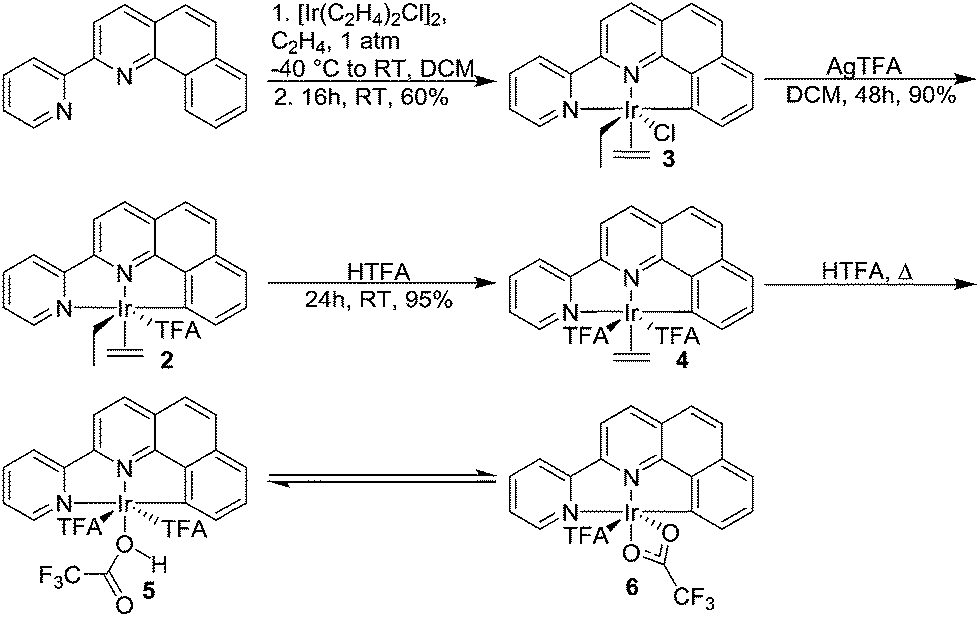
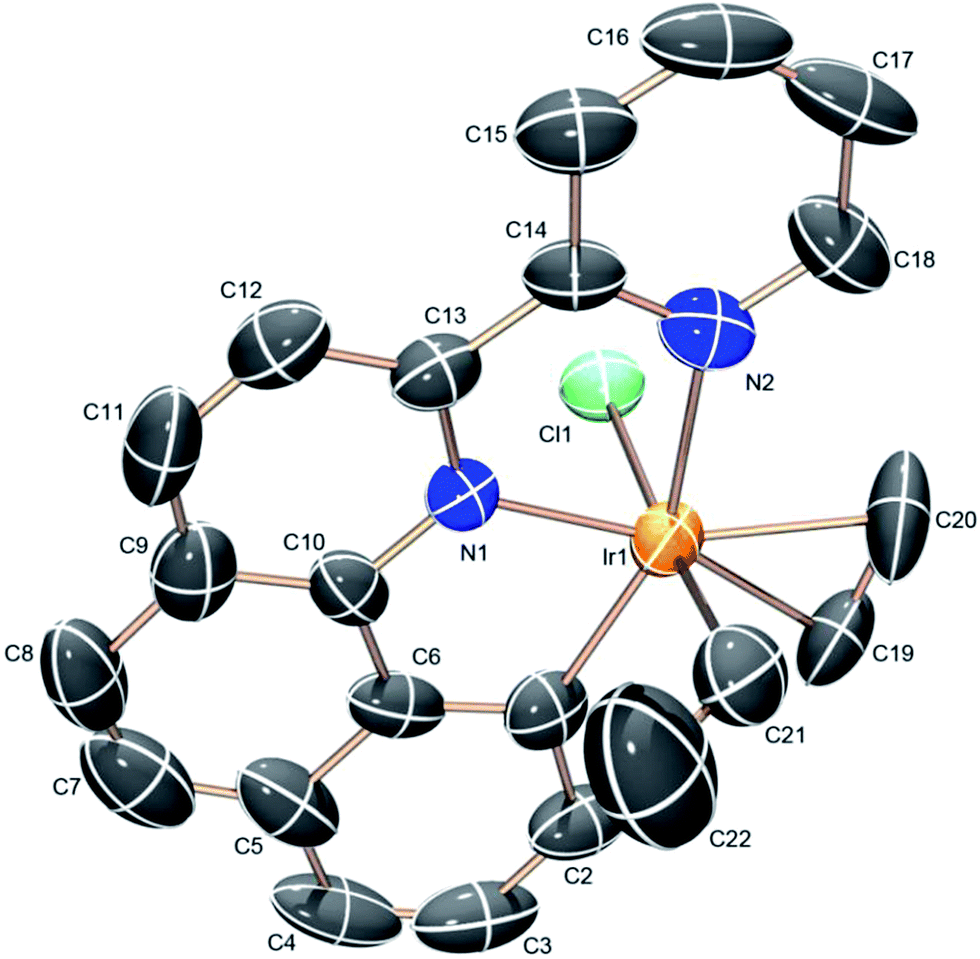
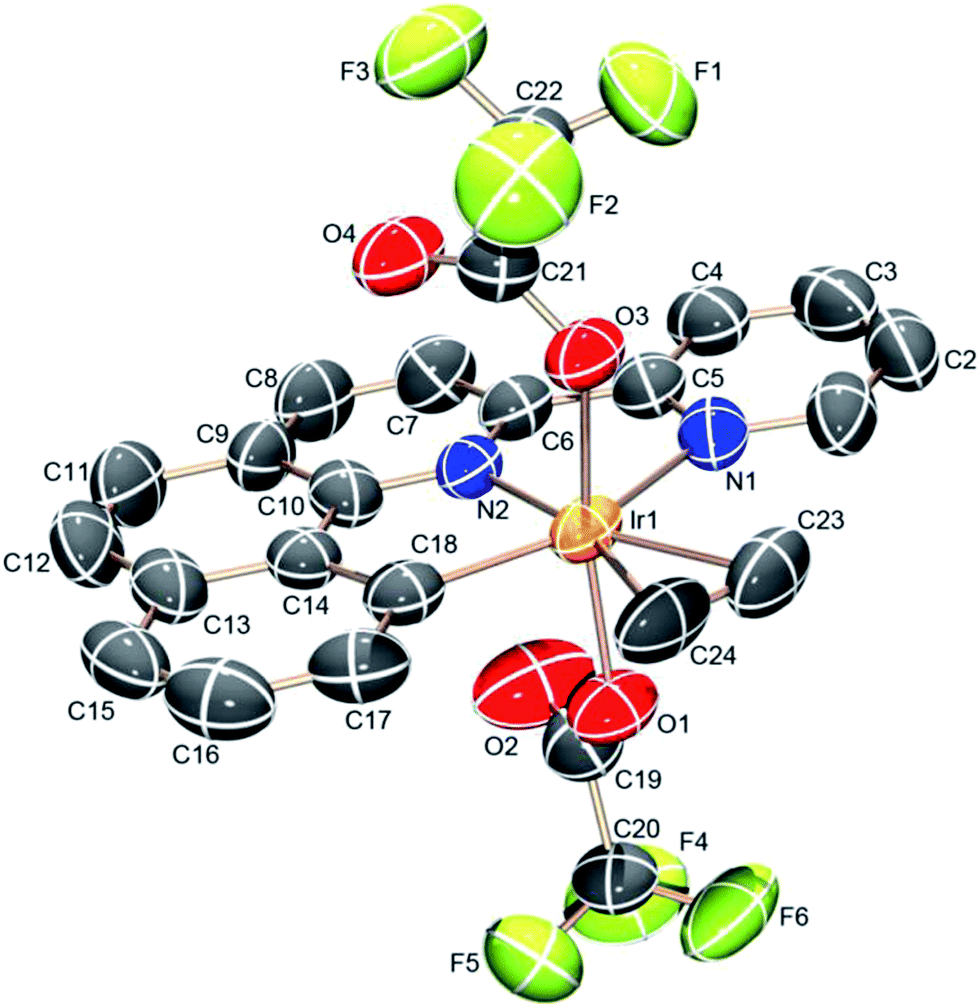
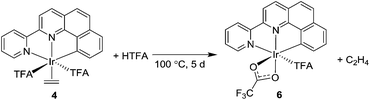
The (NNC-tb)Ir motif is readily accessible by following a similar synthetic methodology to those previously reported (Scheme 3).47 Warming a methylene chloride solution of [Ir(COE)2Cl]2 with 1 eq. of NNC-tb ligand51 from −50 °C under an ethylene atmosphere, followed by stirring overnight at RT provided the six coordinate, IrIII complex, (NNC-tb)Ir(Et)(C2H4)(Cl) (3) as an orange solid in 60% yield over two steps. The freely rotating ethylene ligand is observed as a singlet at δ = 4.1 ppm in the 1H-NMR. Replacement of the chloride is achieved by treatment with AgTFA and stirring at RT for 48 h to give (NNC-tb)Ir(Et)(C2H4)(TFA) (2). Finally, the ethyl group can be removed as ethane by stirring for 24 h at RT in neat HTFA to give (NNC-tb)Ir(C2H4)(TFA)2 (4). All compounds were characterized by multinuclear NMR (1H, 13C, and 19F), HR-MS, elemental analysis, and X-ray crystallography (Fig. 2, for 3, Fig. 3, for 2, and Fig. 4 for 4, respectively). Based on previous work with 1 in our group, heating 2 or 4 in hot HTFA should lead to loss of ethane and ethylene and generate a mixture of 5 and 6 under catalytic conditions (Scheme 3).
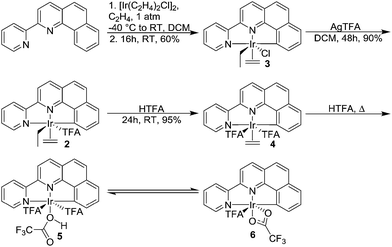 |
| | Scheme 3 Synthesis of (NNC-tb)Ir(R)(L)(X) compounds 3–6. | |
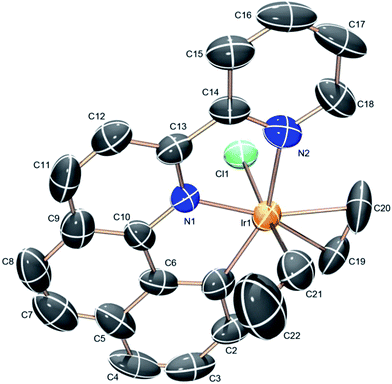 |
| | Fig. 2 ORTEP structure of (NNC-tb)Ir(Et)(C2H4)(Cl) (3) with ellipsoids at 50%. | |
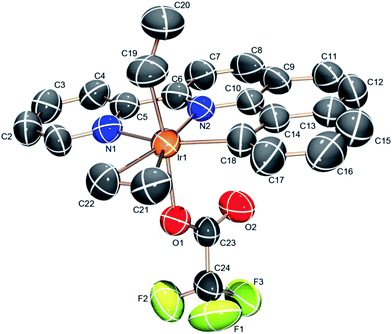 |
| | Fig. 3 ORTEP structure of (NNC-tb)Ir(Et)(C2H4)(TFA) (2) with ellipsoids at 50%. | |
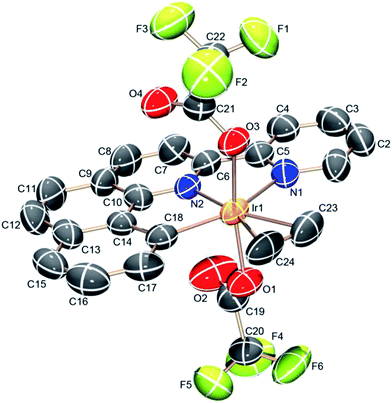 |
| | Fig. 4 ORTEP structure of (NNC-tb)Ir(C2H4)(TFA)2 (4) with ellipsoids at 50%. | |
Analysis of the crystal structures for complexes 2–4 revealed increasing electrophilic character leading to a tightly bound ethylene as the electron-withdrawing TFA groups are added. Substitution of the Cl for TFA leads to an increase in the electrophilicity at the metal center as evidenced by the decreased C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length (Table 1). Back donation from filled Ir π-orbitals would be expected to increase the CC bond length. The observed decrease in CC bond length in moving from complex 3 to the more electron deficient complex, 2, is consistent with less back-bonding and more characteristic of a bound ethylene rather than a metalocyclopropane. In all cases, the bound ethylene is in plane with the NNC ligand in the crystal structures. The CC bond length of coordinated ethylene in complex 3 is 1.386 (16) Å, slightly longer than the CC distance of 1.335 Å of free ethylene52 and shorter than a typical C–C single bond (1.54 Å). The bond distance is more closely resemblant of a delocalized C–C bond, such as that of benzene (1.394 Å). The CC bond distances in complexes 2 and 4 are 1.377 (13) and 1.341 (14) Å, respectively. Successive substitution of the TFA ligand on the iridium center results in a decrease of the CC bond length across a range of ∼0.045 Å. The structural data is consistent with an appreciable electrophilic iridium center and a coordinated ethylene, which is consistent with solution NMR data for complexes 2–4. The Ir–C distances are not particularly sensitive to changes in ligands on Ir and vary from 2.163 (9) to 2.188 (8) Å (Table 1).
C bond length (Table 1). Back donation from filled Ir π-orbitals would be expected to increase the CC bond length. The observed decrease in CC bond length in moving from complex 3 to the more electron deficient complex, 2, is consistent with less back-bonding and more characteristic of a bound ethylene rather than a metalocyclopropane. In all cases, the bound ethylene is in plane with the NNC ligand in the crystal structures. The CC bond length of coordinated ethylene in complex 3 is 1.386 (16) Å, slightly longer than the CC distance of 1.335 Å of free ethylene52 and shorter than a typical C–C single bond (1.54 Å). The bond distance is more closely resemblant of a delocalized C–C bond, such as that of benzene (1.394 Å). The CC bond distances in complexes 2 and 4 are 1.377 (13) and 1.341 (14) Å, respectively. Successive substitution of the TFA ligand on the iridium center results in a decrease of the CC bond length across a range of ∼0.045 Å. The structural data is consistent with an appreciable electrophilic iridium center and a coordinated ethylene, which is consistent with solution NMR data for complexes 2–4. The Ir–C distances are not particularly sensitive to changes in ligands on Ir and vary from 2.163 (9) to 2.188 (8) Å (Table 1).
Table 1 Comparison of the CC and Ir–C bond lengths (Å)
| Entry |
Complex |
CC |
Ir–C |
| 1 |
Ethylene |
1.335 |
— |
| 2 |
(NNC-tb)Ir(Et)(C2H4)(Cl) (3) |
1.386 (16) |
2.179 (10), 2.180 (10) |
| 3 |
(NNC-tb)Ir(Et)(C2H4)(TFA) (2) |
1.377 (13) |
2.163 (9), 2.172 (8) |
| 4 |
(NNC-tb)Ir(C2H4)(TFA)2 (4) |
1.341 (14) |
2.182 (8), 2.188 (8) |
Complex 2 was then examined for the CH activation reaction with a variety of hydrocarbons. H/D exchange reactions between hydrocarbons and deuterated DTFA revealed the system is active for aromatic sp2 C–H bonds (Fig. 5). Heating a 0.5 M solution of benzene in DTFA containing 5 mM of 2 at 150 °C for 3 h followed by GC-MS analysis showed 25% of the CH bonds had been converted to CD bonds (Table 2). With these positive results, reactions with benzylic and aliphatic CH bonds were examined (Fig. 5). For example, heating a stirred solution of DTFA containing 5 mM 2 for 2 h at 180 °C under 500 psi of CH4 led to H/D exchange between methane and the DTFA solvent. Analysis of the gas phase of the reaction mixture indicated that ∼5% of the CH4 was converted to 5.1% CH3D, 0.1%, CH2D2, 0.0% CHD3, and 0.0% CD4. In comparison, 1 was shown to give ∼30% conversion of CH4 to 21.2% CH3D, 6.6% CH2D2, 1.3% CHD3, and 0.6% CD4, under comparable conditions. It is important to note that the reaction mixtures remained completely homogeneous post reaction, in all cases, suggesting that catalyst decomposition is unlikely. Additionally, control reactions show that no background H/D exchange is observed between solvent and methane in the absence of either catalyst. This would suggest that the H/D exchange is the result of reversible CH activation. Thus, the generation of (NNC-r)Ir(CH3)X2 intermediates is plausible, where X is TFA or HTFA.
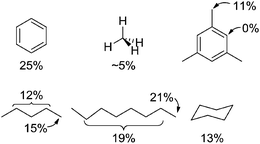 |
| | Fig. 5 H/D exchange results using 2 in DTFA. | |
Table 2 H/D exchange of benzene and DTFA at 150 °C for 3 h catalyzed by 2
| Run |
C6H6 |
C6H5D |
C6H4D2 |
C6H3D3 |
C6H2D4 |
C6H1D5 |
C6D6 |
TOF (×10−2 s−1) |
TON |
| Background no catalyst |
95.2 |
4.6 |
0.1 |
0.0 |
0.0 |
0.0 |
0.0 |
— |
7 |
| 1 |
40.5 |
11.0 |
6.4 |
6.7 |
9.1 |
13.7 |
12.6 |
2.80 |
302 |
| 2 |
43.2 |
10.5 |
5.9 |
6.0 |
8.1 |
13.1 |
13.2 |
2.72 |
293 |
In addition to methane, complex 2 also catalyzes H/D exchange with other alkanes and arenes such as n-octane, cyclohexane, mesitylene, and n-pentane in trifluoroacetic acid based on NMR and GC-MS analysis. With n-octane and n-pentane, exchange is observed at all positions with a small preference for the primary position. This is not surprising, as activation of stronger, primary CH bonds is consistent with other reports on CH activation systems.53–55 This selectivity can be attributed to the stronger M–C bonds formed from scission of primary, sp3 CH bonds as a main driving force for the observed regioselectivity. Secondary CH bonds lead to a weaker M–C bond, which disfavors formation of the internal M–C intermediate. Furthermore, these results are also persuasive evidence against an exchange mechanism involving radicals. The lack of observed H/D exchange on the sp2 CH bond of mesitylene is the result of an extraordinarily fast background reaction between DTFA solvent and mesitylene (likely the result of electrophilic aromatic substitution reactions with the methyl groups stabilizing the carbocation intermediates). This extraordinarily fast background reaction makes further H/D exchange at this position difficult to detect. In comparison to 1, the observed rates of H/D exchange catalyzed by 2 are typically equivalent within error for benzylic CH bonds and slower for aliphatic hydrocarbons. A possible reason for this disparity will be discussed later in this manuscript.
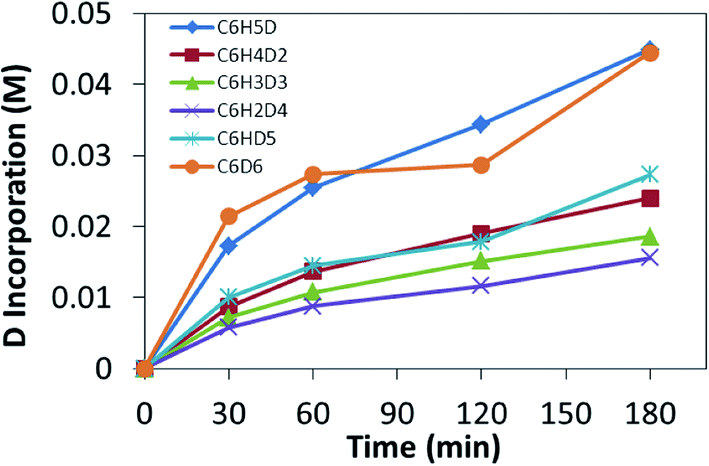
Recently, we utilized a method that allows for distinguishing between rate limiting steps of CH bond coordination or cleavage.56 If the system was following typical Schultz–Flory behavior, C6D1H5 should build up followed by gradual increase in C6D2H4. Given that higher isotopologues of benzene are observed, it would suggest that multiple CH activation events are occurring. H/D exchange reactions between C6H6 and DTFA were monitored by sampling the liquid phase by GC-MS analysis after neutralization of the acid and extraction with diethyl ether. Heating 5 mM solutions of 2 in a 0.5 M solution of benzene in DTFA and sampling the reaction over 3 h led to parallel incorporation of deuterium (Fig. 6). The parallel incorporation of deuterium is suggestive of CH bond coordination as the rate determining step for the CH activation reaction of benzene and can be modeled by the hypothetical energy diagram shown in Fig. 7.
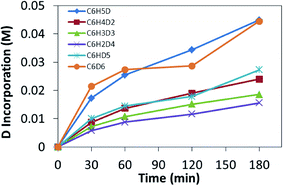 |
| | Fig. 6 Benzene H/D exchange catalyzed by 2 monitored over 3 h at 100 °C. | |
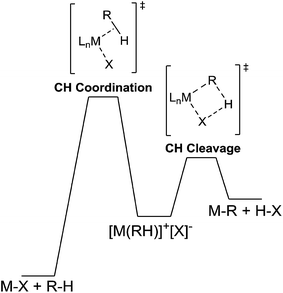 |
| | Fig. 7 Representative energy diagram for CH activation that leads to parallel incorporation. | |
In moving to the “tied back”, NNC-tb ligand, we expected the system to become more stable to the reaction conditions. We found it perplexing, therefore, that our new complex, 2, exhibited less activity for H/D exchange with aliphatic hydrocarbons than did the parent complex, 1. This led us to question the lability of the bound ethylene in 2. However, to avoid any complications by the inclusion of the ethyl ligand, we choose to use complex 4 instead of 2 to examine the lability of only ethylene. Previously, it was reported that NNC ligated Ir complexes underwent facile loss of ethylene under similar conditions.50,57,58 The loss of ethylene from 4 (eqn (2)) was monitored by preparing a 15 mM solution in HTFA in a sealed J-Young NMR tube under argon. The NMR tube contained a coaxial insert containing 1,3,5-trimethoxybenzene in DMSO-D6. The NMR tube was heated at 100 °C and monitored over 5 days by 1H NMR (see ESI†). Monitoring of the ethylene signal at 4.9 ppm revealed that loss of the ethylene from 4 was in fact much slower than in previously developed (tBu-NNC)Ir complexes. A T1/2 = ∼1000 min was observed for the loss of the Ir–ethylene signal.
| |
 | (2) |
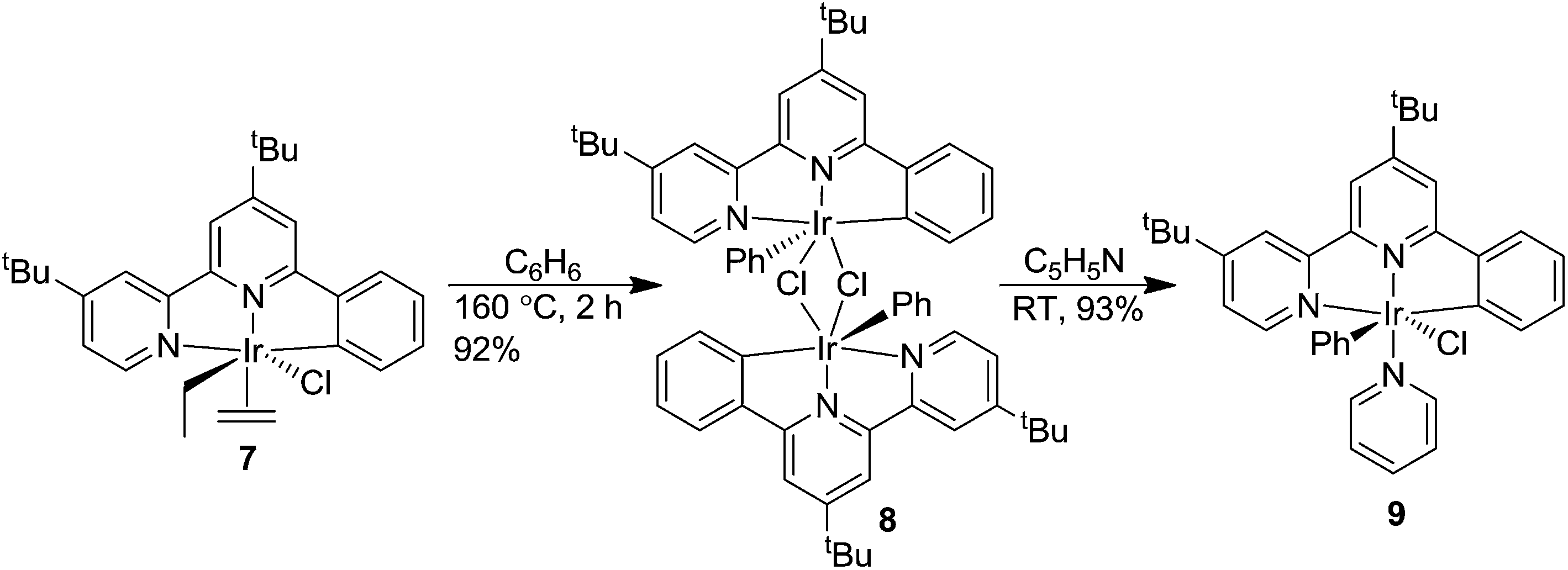
In an effort to improve catalytic CH activation in this system, we designed Ir complexes that were based on the NNC-tb ligand, but did not have a bound ethylene. Previous work in our group has shown that heating NNC ligated Ir complexes (7) in neat hydrocarbon such as benzene led to a bridging Ir dinuclear species (8).59 Treatment of the chloride dinuclear species, 8, with a coordinating amine base such as pyridine gave rise to the formation of a mononuclear complex (tBu-NNC)Ir(Ph)(Py)(Cl) (9, Scheme 4). Following a similar methodology, 3 was heated in neat benzene at 160 °C for 6 h in a sealed glass vessel under vacuum. Removal of the solvent and treatment of the residue with neat 4-methylpyridine (4-MePy) for 2 days, followed by flash column chromatography gave (NNC-tb)Ir(Ph)(4-MePy)(Cl) (10) in 37% yield over two steps as a brown powder. The use of unsubstituted pyridine led to a mixture of products that proved difficult to resolve by chromatography or crystallization. Specifically, 1H NMR interpretations were challenging due to the nature of 21 aryl protons with 17 of them being chemically distinct and overlapping. We found that addition of the methyl group on the pyridine in the 4-position provided a means of identifying pyridine ligated Ir complexes where the methyl group is used as a reference point. Furthermore, the increased solubility of the 4-methylpyridine complex in organic solvents makes further manipulations simpler. Following isolation of 10, stirring in DCM with 4 eq. of AgTFA gave (NNC-tb)Ir(Ph)(4-MePy)(TFA) (11) in quantitative yield (Scheme 5).
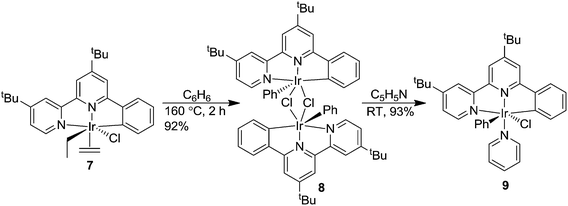 |
| | Scheme 4 Synthesis of (tBu-NNC)Ir(Ph)(Py)(Cl) (9).59 | |
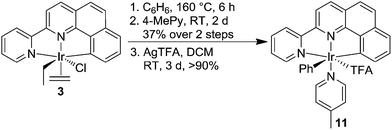 |
| | Scheme 5 Synthesis of (NNC-tb)Ir(Ph)(4-MePy)(TFA) (11). | |
We then set out to determine if 11 was an effective catalyst for CH activation of aromatic substrates. Our group's previous work suggested that the Ir–Ph intermediates of the parent Ir(tBu-NNC) complex 1, 8 or 9, show similar catalytic rates to the parent compounds within experimental error. It was our hope, in moving to the NNC-tb ligated systems, that 11 would be much more active than 2, due to the slower loss of ethylene in the NNC-tb ligated iridium systems than those previously reported for tBu-NNC ligated iridium systems. Therefore, we heated solutions of 11 under identical conditions to those reported for 2 (150 °C, 3 h) and found that the Ir–Ph complex led to significantly increased rates of H/D exchange between C6H6 and DTFA. The rate of H/D exchange was so fast that reactions did not remain pseudo-first-order with respect to benzene, preventing extraction of suitable rate data. The extraordinarily high rates of H/D exchange using 11 prompted us to compare these new complexes to some of our previously reported active CH activation catalysts.47,50 Our goal in development of the “tied-back” NNC was to improve stability (and hopefully enhance catalytic rates) of the parent NNC complex. Pseudo-first-order conditions for H/D exchange between benzene and DTFA catalyzed by 11 were realized by running the reaction at 75 °C for 1 h. In order to ensure that the phenyl substituent on the catalyst did not significantly contribute to the observed H/D exchange, the concentration of benzene in solution was increased to 5 M and the catalyst concentration remained at 5 mM. Control studies run with 11 in DTFA indicate that deuteration of the phenyl substituent occurs rapidly, even at room temperature, and that deuterium is incorporated into the resultant benzene (see ESI†). Catalysts 1, 2, and 11 were screened under identical conditions in order to make a direct comparison of their activity. In all three cases, the reaction mixtures remained homogeneous post reaction, suggesting that catalyst decomposition is not occurring. The results of this study indicate that 11 is approximately an order of magnitude faster than 1 and 2 (Table 3). Interestingly, there are no observed differences in rate between the “tied-back” and parent NNC complexes within experimental error. This would suggest a common mechanism for CH activation between 1 and 2. The higher observed rate for 11 correlates well with a more tightly bound ethylene in 2 than had been expected and it is likely that our increase in catalytic activity, when using 11, results from the generation of a highly active Ir cation possessing open coordination sites that are not readily accessible when ethylene is present in the complex, especially at lower temperatures. Thus, slow loss of ethylene from 2 results in moderate catalyst inhibition at the start of reactions.
Table 3 Comparison of various Ir–NNC catalyst CH activation systemsa
|
| Catalyst |
TON |
TOF (×10−3) |
| Conditions: 5 mM catalyst, 5 M C6H6 in DTFA, at 75 °C under Argon and analyzed by GC-MS after 1 h. |
| 1 |
7.5 ± 0.7 |
2.0 ± 0.1 |
| 2 |
8 ± 2 |
2.5 ± 0.7 |
| 11 |
90 ± 16 |
25 ± 4 |
Given that both 2 and 11 were competent for CH activation of benzene, we examined them for catalytic the oxy-functionalization of benzene. Catalytic functionalization was attempted using either 2 or 11 with an assortment of one and two – electron oxidants. We had particularly been interested in the use of air – regenerable oxidants, such as CuII, with the idea of using Wacker – like conditions for hydrocarbon oxidation. Unfortunately, 2 did not exhibit increased activity for benzene functionalization when compared to 1 and a maximum TON of 6.9 was achieved (see ESI†). Interestingly, 11 did not show any activity for benzene functionalization although it was highly active for CH activation. Attempts to stoichiometrically functionalize the phenyl substituent on 11 resulted in the formation of benzene resulting from protonolysis of the phenyl substituent. Catalytic runs were also unsuccessful. Although no TON was observed with 11 for the oxy-functionalization of benzene, it is possible that the 4-MePy substituent is interfering with the functionalization. Further study is needed to elucidate the details of this system.
Conclusion
In our efforts to design catalysts for hydrocarbon oxidation in weakly acidic media several new Ir complexes based on the use of a cyclometalated NNC ligand have been synthesized and examined for reactions with hydrocarbons. Contrary to our expectations, ligating Ir with a “tied back” version of the NNC ligand (see complex 2) relative to the original “non-tied back”complex did not result in increased rates of H/D exchange. Our studies have indicated that the lower rate of exchange with aliphatic substrates, such as methane, may be due to a more tightly bound ethylene in the precursor to the active catalyst and not from catalyst decomposition. The slow loss of ethylene from 2 may inhibit entrance into the catalytic cycle from the iridium pre-catalyst as two cis-coordination sites are required for CH activation. Replacement of the ethylene ligand with 4-MePy (see complex 11) resulted in substantially increased rates of H/D exchange with benzene. Importantly, the use of oxidants, such as CuII, gave oxy-functionalized aryl products of benzene as either phenol or phenyltrifluoroester using complex 2. However, the yields are low and there is a background reaction facilitated by Cu in HTFA under the reactions conditions. Unfortunately, complex 11 gave only trace amounts of oxy-functionalized products and further investigation is needed to elucidate the reason for this. Further investigations of this reaction with iridium and other metals are ongoing in our lab.
Experimental Section
General considerations
All manipulations were carried out using an argon filled MBraun glovebox and standard Schlenk techniques using oven dried glassware (>1 h at 110 °C under vacuum, −30 mm Hg). Methylenechloride was HPLC grade from Fisher and distilled from P2O5 under argon prior to use. Benzene was HPLC grade from EMD and distilled from Na/benzophenone under argon prior to use. Pentane was HPLC grade from EMD and distilled over Na/benzophenone under argon prior to use. Reagent-grade chemicals and solvents were purchased from Sigma Aldrich or Alfa Aesar and used as is unless otherwise specified. Deuterated solvents were purchased from Cambridge Isotope Inc. and degassed and stored under argon before use. Deuterated methylenechloride was further stored over 4 Å molecular sieves. Deuterated trifluoroacetic acid was degassed before use by several freeze–pump–thaw cycles and stored under argon. Trifluoroacetic acid was HPLC grade from Fisher and degassed before use by several freeze–pump–thaw cycles and stored under argon. IrCl3·(H2O)x was obtained from Pressure Chemical. Elemental Analyses were performed by Columbia Analytical Services; Tucson, Arizona or Atlantic Microlab, Inc. of Norcross, Georgia. Electrospray Ionization (ESI) mass spectroscopy was performed at the University of Illinois Mass Spec Facility; Urbana, Illinois. Liquid phase organic products were analyzed with a Shimadzu GC-MS QP2010S equipped with cross-linked methyl silicone gum capillary column, RTX-5. Gas measurements were performed using an Agilent GasPro column. The retention times of the products were confirmed by known standards. NMR spectra were obtained on a Bruker Digital Avance III 400 (400.132 MHz for 1H, 100.623 MHz for 13C, and 376.461 MHz for 19F) spectrometer. Chemical shifts are given in ppm relative to residual solvent proton resonances or to a stated internal or external standard. Liquid phase flash chromatography was performed on a Teledyne Isco Combiflash Rf and all solvents were of HPLC grade and obtained from Fisher or EMD. X-ray crystallography was performed at the Center for Nanostructured Materials located at the University of Texas at Arlington.
Benzo[h]quinoline n-oxide
A solution of benzo[h]quinoline (1.8 g, 10.0 mmol) dissolved in 40 mL of CHCl3 was cooled in an ice bath. To this cooled solution, MCPBA (3.0 g, 15.0 mmol) in 60 mL of CHCl3 was added and the reaction stirred for 4 days at RT. The solution was washed with K2CO3, dried with MgSO4, and concentrated in vacuo. The residue was washed with diethyl ether to give a crystalline solid in 65–70% yield. Characterization of the compound corresponded to that previously reported.60
2-Chlorobenzo[h]quinoline
A mixture of benzo[h]quinoline n-oxide (13.8 g, 71 mmol) in 60 mL of POCl3 was refluxed for 30 min. The cooled reaction mixture was poured into crushed ice, neutralized with concentrated NH4OH solution, and extracted with CH2Cl2 (3 × 50 mL). The combined extracts were washed with water, dried over MgSO4, and evaporated under reduced pressure. The residue was flash-chromatographed on silica with a gradient from hexanes to CH2Cl2 as the eluent to give 2-chlorobenzo[h]quinoline (44–48%) as the first major fraction. Characterization of the compound corresponded to that previously reported.60
2-Bromobenzo[h]quinoline
The reaction was performed in a well dried round bottom flask equipped with a reflux condenser under an argon atmosphere. To a solution of 2-chlorobenzo[h]quinoline in acetic acid (60 mL) kept under reflux, ∼10 mL of 48% HBr was added slowly. The reaction was then refluxed for 6 h. The excess of solvent was removed in vacuo and the resulting product washed with a NaHCO3 saturated aqueous solution, extracted into CH2Cl2, and dried over MgSO4. The residue after removing the solvent was flash-chromatographed on silica with a gradient from hexanes to CH2Cl2 as the eluent to yield 55–68% of 2-bromobenzo[h]quinoline. Characterization of the compound corresponded to that previously reported.61
2-(2′-Pyridyl)-benzo[h]quinoline
A mixture of 2-bromobenzo[h]quinolone (1.9 g 7.36 mmol), 2-(tri-nbutylstannyl) pyridine (1.2 eq., 3.76 g, 8.83 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.1 eq., 850 mg, 0.74 mmol) was loaded into an oven dry Schlenk flask with a reflux condenser.62 The solids were dissolved in dry toluene (60 mL) and refluxed under argon for 2 days. After cooling to room temperature, water was added and the mixture was concentrated under reduced pressure. Flash chromatography of the residue on silica gel, eluting with hexanes, followed by EtOAc/hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) gave the product (NNC-tb) as a white/yellow solid (1.7 g, 90%). Characterization of the compound corresponded to that previously reported.63
:3) gave the product (NNC-tb) as a white/yellow solid (1.7 g, 90%). Characterization of the compound corresponded to that previously reported.63
(NNC-tb)Ir(Et)(C2H4)(Cl) (3)
A Schlenk bomb was loaded with 3 g (3.35 mmol) of [Ir(COE)2Cl]2 and then dissolved in dry/degassed pentane in the glovebox. The flask was cooled to −50 °C under argon and ethylene was bubbled through for 30 min followed by slow warming to RT. The reaction was then cooled to −50 °C and transferred by cannula to a precooled (−50 °C) Schlenk frit and washed with cold pentane (3 × 50 mL) to remove COE while under continuous ethylene pressure. The receiving flask was then changed and the system washed at RT with dry CH2Cl2 (3 × 30 mL) all while under ethylene pressure. To the collected filtrate solution at −50 °C, 1.72 g (2.23 mmol) of NNC-tb dissolved in 45 mL of dry CH2Cl2 was cannula transferred in. The flask was then warmed to RT and stirred for 16 h to yield a red/orange solution with a precipitate, which was evacuated to dryness on a high vac line. The reaction was then filtered over a 1 × 3′′ plug of silica gel first washing with ether, then EtOAc, followed by EtOAc with 10% methanol to elute the product as an orange band. There is a red/brown band that elutes with EtOAc as well as ligand/COE with diethyl ether that may be discarded. The orange residue was dissolved in DCM and crashed out in stirring pentane followed by filtration on a sintered glass frit to give a 65% yield of (NNC-tb)Ir(Et)(C2H4)(Cl). 1H NMR (CD2Cl2, 400 MHz): δ 9.35 (d, J = 5.1 Hz, 1H), 8.24 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 8.01 (t, J = 7.8 Hz, 1H), 7.91 (d, J = 8.9 Hz, 1H), 7.85 (d, J = 6.8 Hz, 1H), 7.62 (m, 4H), 4.1 (s, 4H, C2H4), 0.41 (m, 1H, Ir–CH2–CH3), 0.25 (m, 1H, Ir–CH2–CH3), −0.44 (t, J = 7.6 Hz, 3H, Ir–CH2–CH3). 13C NMR {1H} (CD2Cl2, 100 MHz): δ 163.9, 163.0, 162.3, 158.4, 153.2, 151.2, 144.8, 144.4, 135.0, 131.7, 124.9, 124.6, 122.7, 119.6, 116.2, 115.1, 65.9. HR-MS Q-Tof calc. for C20H17N2Ir [M + H–Cl–C2H4]+: 478.1021; found: 478.1020. Anal. Calc. for C28H36ClIrN2: C 53.53; H 5.78; N 4.46, Cl 5.64; found: C 53.44, H 5.67, N 4.39, Cl 5.59. X-ray: crystals were obtained by crystallization from a CH2Cl2 solution at −20 °C.
(NNC-tb)Ir(Et)(C2H4)(TFA) (2)
In a 30 mL amberized vial with a magnetic stir bar, 250 mg of (NNC-tb)Ir-(Et)(C2H4)(Cl) and 102 mg (1.1 eq.) of AgTFA was dissolved in 25 mL of dry DCM. The reaction was stirred in the dark for 2 days and then filtered over celite and evacuated to dryness to afford an orange-red powder. The compound was recrystallized from DCM/Pentane at −20 °C to yield 76% of (NNC-tb)Ir(Et)(C2H4)(TFA). 1H NMR (CD2Cl2, 400 MHz): δ 9.42 (d, J = 5.6 Hz, 1H), 8.32 (d, J = 8.3 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.12 (m, 2H), 7.96 (d, J = 8.5 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.67 (m, 4H), 4.1 (m, J = 19 Hz, 4H, C2H4), 0.51 (m, 1H, Ir–CH2–CH3), 0.36 (m, 1H, Ir–CH2–CH3), −0.48 (t, J = 7.6 Hz, 3H, Ir–CH2–CH3). 13C NMR {1H} (CD2Cl2, 100 MHz): δ 160.00, 153.32, 152.88, 143.39, 140.38, 139.76, 137.08, 135.47, 132.18, 131.80, 131.68, 127.82, 126.92, 123.55, 123.50, 121.35, 117.28, 111.84, 66.68 (s, Ir–C2H4), 14.83 (s, Ir–CH2CH3), and −14.17 (s, Ir–CH2CH3). The carbons from the –OC(O)CF3 group were not observed. 19F NMR {1H} (CD2Cl2, ref. to CFCl3, 376 MHz): δ −75.77. HR-MS Q-Tof calc. for C22H16N2O2F3Ir [M + H+–Et]+: 590.0793; found: 590.0786. Elemental Analysis Calc. for C30H36F3IrN2O2: C 51.05; H 5.14; F 8.07; N 3.97; found C 50.75; H 4.98; F 8.14; N 3.96. X-ray: crystals were obtained by crystallization from a CH2Cl2/ether solution at −20 °C.
(NNC-tb)Ir(C2H4)(TFA)2 (4)
In a 100 mL RBF, 232 mg of (NNC-tb)Ir-(Et)(C2H4)(TFA) was dissolved in 25 mL of HTFA. The reaction was stirred for 1 day and evacuated to dryness to give a yellow powder. The compound was purified by flash column chromatography on silica gel using a 1:1:1 mixture of hexanes:DCM:EtOAc to give 77% of (NNC-tb)Ir(C2H4)(TFA)2. 1H NMR (CD2Cl2, 400 MHz): δ 9.53 (d, J = 5.3 Hz, 1H), 8.42 (d, J = 8.3 Hz, 1H), 8.37 (d, J = 7.8 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H), 8.20 (dt, J = 7.9, 1.5 Hz, 1H), 8.01 (d, J = 8.9 Hz, 1H), 7.91 (dd, J = 6.1, 2.3 Hz, 1H), 7.76 (m, 4H), 5.02 (s, 4H, C2H4). 13C NMR (CD2Cl2, 100 MHz): δ 163.8 (q, J = 36.1 Hz), 160.8, 154.8, 154.1, 153.9, 143.9, 141.0, 138.9, 135.4, 133.4, 131.9, 131.3, 128.9, 128.1, 127.2, 123.8 (t, J = 6.3 Hz), 117.7, 112.0 (q, J = 290.3 Hz), 73.1. 19F NMR {1H} (CD2Cl2, ref. to CFCl3, 376 MHz): δ −75.60. HR-MS Q-Tof calc. for C24H16N2O4F6Ir [M + H]+: 703.0644; found: 703.0638. Anal. Calc. for C30H31F6IrN2O4·H2O: C 44.61; H 4.12; N 3.47, found C 44.73, H 4.06, N 3.46. X-ray: crystals were obtained by crystallization from a CH2Cl2/n-hexane (1:1) solution at −20 °C.
(NNC-tb)Ir(Ph)(4-methylpyridine)(Cl) (10)
In a thick-walled Schlenk bomb containing a magnetic stir bar and a resealable PTFE valve, 300 mg (0.55 mmol) of (NNC-tb)Ir-(Et)(C2H4)(Cl) was dissolved in 100 mL of dry benzene under argon. The red solution was then frozen in liq. N2 and the head space was evacuated. The flask was thawed and then heated at 160 °C for 6 h in a well stirred oil bath. During the course of the reaction and upon cooling, a dark brown solid precipitated from solution. The solvent was then removed under vacuum to give a dark colored residue. In a 250 mL RBF, the residue was dissolved in 30 mL of 4-methylpyridine. The reaction was stirred for 2 days under air at RT. The solvent was then removed by rotary evaporation to give a dark brown residue. Flash chromatography on silica eluting with Hexanes to neat THF yielded the product as the second dark brown band in 37% yield over two steps. 1H NMR (CD2Cl2, 400 MHz): δ 9.29 (d, J = 5.0 Hz, 1H), 8.12 (t, J = 3.9 Hz, 1H), 8.08 (d, J = 8.9 Hz, 1H), 8.04 (d, J = 6.3 Hz, 2H), 7.98 (dt, J = 7.8, 1.5 Hz, 1H), 7.91 (q, J = 21.6, 8.6 Hz, 2H), 7.84 (d, J = 8.8 Hz, 1H), 7.68 (dt, J = 5.3, 1.0 Hz, 1H), 7.55 (m, 3H), 6.72 (d, J = 5.6 Hz, 4H), 6.42 (m, 3H), 2.09 (s, Ir–C5H4N–CH3, 1H). 13C NMR (CD2Cl2, 100 MHz): δ 158.6, 158.5, 155.2, 150.9, 150.4, 150.1, 149.3, 146.6, 138.9, 136.7, 136.7, 135.8, 134.8, 132.5, 132.3, 131.9, 128.7, 128.1, 126.2, 125.8, 125.6, 123.3, 123.2, 121.9, 118.2, 117.0, 21.1 (Ir–C5H4N–CH3). HR-MS Q-Tof calc. for C30H24N3IrCl [M + H]+: 654.1288; found: 654.1296. Anal. Calc. for C30H23ClIrN3: C 55.16, H 3.55, N 6.43; found, C 54.98, H 3.42, N 6.45.
(NNC-tb)Ir(Ph)(4-methylpyridine)(TFA) (11)
In a 100 mL RBF, 150 mg of (NNC-tb)Ir-Ph 4MePy Cl and 202 mg (4 eq.) of AgTFA were dissolved in 80 mL of dry DCM. The reaction was stirred in the dark for 3 days. The sample was then filtered over a plug of alumina with a celite bottom and washed with DCM and then DCM/10% MeOH to elute a yellow/red band. The solution was then evacuated to dryness. The residue was then recrystallized from DCM/pentane at −20 °C to give >90% yield of the title product. Alternatively, the residue can be sonicated in diethyl ether (3 × 30 mL) followed by centrifugation and decanting of the liquid to reisolate the residue. This procedure affords purity levels of that achieved by recrystallization. 1H NMR (CD2Cl2, 400 MHz): δ 10.16 (d, J = 5.4 Hz, 1H), 8.17 (d, J = 7.9 Hz, 1H), 8.01 (m, 3H), 7.93 (d, J = 8.6 Hz, 1H), 7.88 (d, J = 6.4 Hz, 2H), 7.83 (d, J = 8.5 Hz, 1H), 7.65 (dt, J = 7.2, 1.3 Hz, 1H), 7.59 (m, 3H), 6.78 (d, J = 6.1 Hz, 2H), 6.56 (m, 2H), 6.48 (m, 3H), 2.11 (s, Ir–C5H4N–CH3, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 158.58, 157.31, 156.09, 155.71, 149.82, 149.75, 148.29, 145.28, 138.99, 135.50, 135.30, 134.69, 132.08, 131.62, 129.94, 129.36, 129.11, 126.61, 126.02, 125.36, 123.42, 123.18, 122.32, 118.64, 116.90, and 21.15 (Ir–NC5H4–CH3). The carbons from the –OC(O)CF3 group were not observed. 19F NMR {1H} (CD2Cl2, 376 MHz): δ −75.45. HR-MS Q-Tof calc. for C26H18F3IrN3O2 [M–Ph]+: 654.0981; found: 654.654.0987. Anal. Calc. for C32H23F3IrN3O2: C 52.60, H 3.17, N 5.75; found, C 52.31, H 3.26, N 5.83.
General procedure for H/D exchange (gases)
A 4 mL high pressure stainless steel bomb containing a glass liner with a magnetic stir bar was charged with 1.0 mL (1.31 × 10−2 mol) of a CF3COOD solution containing 5 mM catalyst. The flask was heated for 120 min on a temperature controlled aluminum block maintained at 180 °C (unless otherwise mentioned). The gas phase was sampled and analyzed by GC-MS. The percent deuterium incorporation was determined by deconvoluting the mass fragmentation pattern for methane using a Microsoft Excel program.57 An important assumption is built into the program is that there are no isotope effects on the fragmentation pattern of methane. The parent ion peak of methane is relatively stable towards fragmentation and can be used to quantify the exchange reactions. The mass fragmentation pattern from m/z of 12 to 21 was analyzed for each reaction and compared to control reactions not containing catalyst. The results obtained by this method are accurate within ±5% of deuterium incorporation or loss.
General procedure for H/D exchange (liquids)
A 4 mL Schlenk flask with a resealable PTFE valve and a magnetic stir bar was charged with 1.0 mL (1.31 × 10−2 mol) of a CF3COOD solution containing 0.5 M substrate and 5 mM of catalyst (unless otherwise mentioned). The flask was heated for 180 min in a well stirred oil bath maintained at 150 °C for arenes and 180 °C for all other hydrocarbons. The liquid phase was sampled and analyzed by GC-MS and 1H NMR. Liquid hydrocarbon substrates were compared to control reactions not containing catalyst and any background reactions were subtracted accordingly.
The percent deuterium incorporation into benzene when determined by GC-MS was performed by deconvoluting the mass fragmentation pattern of benzene using a Microsoft Excel program.57 An important assumption is built into the program is that there are no isotope effects on the fragmentation pattern of benzene. The parent ion peak of benzene is relatively stable towards fragmentation and can be used to quantify the exchange reactions. The mass fragmentation pattern from m/z of 78 to 84 was analyzed for each reaction and compared to control reactions not containing catalyst. The results obtained by this method are accurate within ±5% of deuterium incorporation or loss. All other hydrocarbons, besides benzene, were determined by 1H NMR comparison of control and catalytic reactions.
H/D exchange over time leads to parallel incorporation of D
Following our general procedures, H/D exchange reactions were sampled at 30, 60, 120, and 180 min and monitored by GC-MS.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
We thank Dr Kenneth Young for helpful discussions regarding the synthesis and characterization of the iridium complexes. This work was supported as part of the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center Funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0001298 (to R.A.P.) and the Scripps Research Institute, Scripps Florida.
References
- G. A. Olah and Á. Molnár, Hydrocarbon Chemistry, Wiley-Interscience, 2nd edn, 2003 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methane Economy, Wiley-VCH, 2006 Search PubMed.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed.
- R. A. Periana, G. Bhalla, W. J. Tenn, K. J. H. Young, X. Y. Liu, O. Mironov, C. J. Jones and V. R. Ziatdinov, J. Mol. Catal. A: Chem., 2004, 220, 7–25 CrossRef CAS PubMed.
- B. L. Conley, W. J. Tenn, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8–23 CrossRef CAS PubMed.
- B. G. Hashiguchi, C. H. Hövelmann, S. M. Bischof, K. S. Lokare, C. H. Leung and R. A. Periana, in Energy Production and Storage: Inorganic Chemical Strategies for a Warming World, Wiley, New York, 2010, p. 150 Search PubMed.
- E. G. Chepaikin, Russ. Chem. Rev., 2011, 80, 363–396 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Loffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340–343 CAS.
- X. Gang, H. Birch, Y. M. Zhu, H. A. Hjuler and N. J. Bjerrum, J. Catal., 2000, 196, 287–292 CrossRef CAS.
- E. S. Rudakov, V. V. Zamashchikov, A. I. Lutsyk and A. P. Yaroshenko, Dokl. Akad. Nauk UzSSR, 1975, 224, 153–156 CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS.
- O. A. Mironov, S. M. Bischof, M. M. Konnick, B. G. Hashiguchi, V. R. Ziatdinov, W. A. Goddard 3rd, M. Ahlquist and R. A. Periana, J. Am. Chem. Soc., 2013, 135, 14644–14658 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS PubMed.
- R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083–4091 CrossRef CAS PubMed.
- N. F. Gol'dshleger, V. V. Es'kova, A. A. Shteinman and A. E. Shilov, Russ. J. Phys. Chem., 1972, 46, 785–786 Search PubMed.
- J. A. Labinger and J. E. Bercaw, Top. Organomet. Chem., 2011, 35, 29–60 CrossRef CAS.
- M. Ahlquist, R. A. Periana and W. A. Goddard, Chem. Commun., 2009, 2373–2375, 10.1039/b821854d.
- V. R. Ziatdinov, J. Oxgaard, O. A. Mironov, K. J. H. Young, W. A. Goddard and R. A. Periana, J. Am. Chem. Soc., 2006, 128, 7404–7405 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- A. V. Pawlikowski, A. D. Getty and K. I. Goldberg, J. Am. Chem. Soc., 2007, 129, 10382–10393 CrossRef CAS PubMed.
- A. N. Vedernikov, S. A. Binfield, P. Y. Zavalij and J. R. Khusnutdinova, J. Am. Chem. Soc., 2006, 128, 82–83 CrossRef CAS PubMed.
- R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. J. Jones, Science, 2003, 301, 814–818 CrossRef CAS PubMed.
- M. Zerella, S. Mukhopadhyay and A. T. Bell, Chem. Commun., 2004, 1948–1949, 10.1039/b405549g.
- S. Chempath and A. T. Bell, J. Am. Chem. Soc., 2006, 128, 4650–4657 CrossRef CAS PubMed.
- E. Gretz, T. F. Oliver and A. Sen, J. Am. Chem. Soc., 1987, 109, 8109–8111 CrossRef CAS.
- L. C. Kao, A. C. Hutson and A. Sen, J. Am. Chem. Soc., 1991, 113, 700–701 CrossRef CAS.
- C. J. Jones, D. Taube, V. R. Ziatdinov, R. A. Periana, R. J. Nielsen, J. Oxgaard and W. A. Goddard, Angew. Chem., Int. Ed., 2004, 43, 4626–4629 CrossRef CAS PubMed.
- D. E. De Vos and B. E. Sels, Angew. Chem., Int. Ed., 2005, 44, 30–32 CrossRef CAS PubMed.
- M. Ahlquist, R. J. Nielsen, R. A. Periana and W. A. Goddard, J. Am. Chem. Soc., 2009, 131, 17110–17115 CrossRef CAS PubMed.
- B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS PubMed.
- B. C. Bales, P. Brown, A. Dehestani and J. M. Mayer, J. Am. Chem. Soc., 2005, 127, 2832–2833 CrossRef CAS PubMed.
- J. M. Mayer, Acc. Chem. Res., 1998, 31, 441–450 CrossRef CAS.
- S. N. Brown and J. M. Mayer, J. Am. Chem. Soc., 1996, 118, 12119–12133 CrossRef CAS.
- Biomimetic Oxidations Catalyzed by Transition Metal Complexes, ed. B. Meunier, Imperial College, London, 2000 Search PubMed.
- G. A. Olah and R. H. Schlosberg, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS.
- G. A. Olah, J. Org. Chem., 2005, 70, 2413–2429 CrossRef CAS PubMed.
- G. A. Olah and G. K. S. Prakash, Superacids, John Wiley and Sons, New York, 1985 Search PubMed.
- W. J. Tenn, K. J. H. Young, G. Bhalla, J. Oxgaard, W. A. Goddard and R. A. Periana, J. Am. Chem. Soc., 2005, 127, 14172–14173 CrossRef CAS PubMed.
- S. M. Kloek, D. M. Heinekey and K. L. Goldberg, Angew. Chem., Int. Ed., 2007, 46, 4736–4738 CrossRef CAS PubMed.
- Y. Feng, M. Lail, K. A. Barakat, T. R. Cundari, T. B. Gunnoe and J. L. Petersen, J. Am. Chem. Soc., 2005, 127, 14174–14175 CrossRef CAS PubMed.
- B. G. Hashiguchi, K. J. H. Young, M. Yousufuddin, W. A. Goddard and R. A. Periana, J. Am. Chem. Soc., 2010, 132, 12542–12545 CrossRef CAS PubMed.
- M. H. Emmert, J. B. Gary, J. M. Villalobos and M. S. Sanford, Angew. Chem., Int. Ed., 2010, 49, 5884–5886 CrossRef CAS PubMed.
- X. Chen, X. S. Hao, C. E. Goodhue and J. Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- Y. H. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed.
- K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- K. J. H. Young, J. Oxgaard, D. H. Ess, S. K. Meier, T. Stewart, W. A. Goddard and R. A. Periana, Chem. Commun., 2009, 3270–3272, 10.1039/b823303a.
- R. P. Muller, D. M. Philipp and W. A. Goddard, Top. Catal., 2003, 23, 81–98 CrossRef CAS.
- X. Xu, J. Kua, R. A. Periana and W. A. Goddard, Organometallics, 2003, 22, 2057–2068 CrossRef CAS.
- S. K. Meier, K. J. H. Young, D. H. Ess, W. J. Tenn, J. Oxgaard, W. A. Goddard and R. A. Periana, Organometallics, 2009, 28, 5293–5304 CrossRef CAS.
- Y. D. Jahng and J. G. Park, Bull. Korean Chem. Soc., 1999, 20, 1200–1204 CAS.
- A. J. Gordon and R. A. Ford, The chemist's companion: a handbook of practical data, techniques, and references, Wiley, New York, 1972 Search PubMed.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7332–7346 CrossRef CAS.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7346–7355 CrossRef CAS.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1984, 106, 7272–7273 CrossRef CAS.
- S. M. Bischof, D. H. Ess, S. K. Meier, J. Oxgaard, R. J. Nielsen, G. Bhalla, W. A. Goddard and R. A. Periana, Organometallics, 2010, 29, 742–756 CrossRef CAS.
- K. J. H. Young, S. K. Meier, J. M. Gonzales, J. Oxgaard, W. A. Goddard and R. A. Periana, Organometallics, 2006, 25, 4734–4737 CrossRef CAS.
- K. J. H. Young, M. Yousufuddin, D. H. Ess and R. A. Periana, Organometallics, 2009, 28, 3395–3406 CrossRef CAS.
- S. K. Meier, Catalytic C–H Activation by Cyclometallated Iridium Hydroxo Complexes in Aqueous Media, Thesis presented to the University of Southern California, 2008.
- A. Cappelli, M. Anzini, S. Vomero, L. Mennuni, F. Makovec, E. Doucet, M. Hamon, G. Bruni, M. R. Romeo, M. C. Menziani, P. G. De Benedetti and T. Langer, J. Med. Chem., 1998, 41, 728–741 CrossRef CAS PubMed.
- W. Baratta, M. Ballico, S. Baldino, G. Chelucci, E. Herdtweck, K. Siega, S. Magnolia and P. Rigo, Chem.–Eur. J., 2008, 14, 9148–9160 CrossRef CAS PubMed.
- C. Barolo, M. K. Nazeeruddin, S. Fantacci, D. Di Censo, P. Comte, P. Liska, G. Viscardi, P. Quagliotto, F. De Angelis, S. Ito and M. Gratzel, Inorg. Chem., 2006, 45, 4642–4653 CrossRef CAS PubMed.
- E. C. Riesgo, Y. Z. Hu, F. Bouvier, R. P. Thummel, D. V. Scaltrito and G. J. Meyer, Inorg. Chem., 2001, 40, 3413–3422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic data, calibration curves for GC data interpretation, labeling studies, oxy-functionalization studies, crystallographic data, and CIF files. CCDC 990488–990490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra06251e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.