
Recent developments in one-pot tandem oxidation process coupling reactions
Abstract
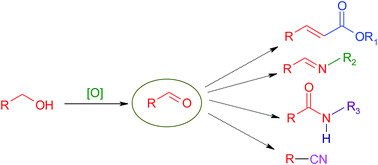
The tandem oxidation process (TOP) constitutes a novel approach to synthetic organic chemistry. This procedure involves the oxidation of the alcohol to the aldehyde which is trapped in situ by a nucleophile leading to a variety of synthetically useful compounds. In recent times, many research groups have focused their attention on developing new approaches to the one-pot coupling process. In this review, we highlight the latest additions, over the past nine years, to the TOP realm such as the use of novel oxidants, innovative catalysts and alternate reaction conditions. In addition, the introduction of TOP to multicomponent reactions and total syntheses will also be discussed.
 Please wait while we load your content...
Please wait while we load your content...

